参考文献
Denti L, Khorsand P, Bonizzoni P, et al. SVDSS: structural variation discovery in hard-to-call genomic regions using sample-specific strings from accurate long reads[J]. Nature Methods, 2023, 20(4): 550-558.
点击观看官方视屏 .
计算原理
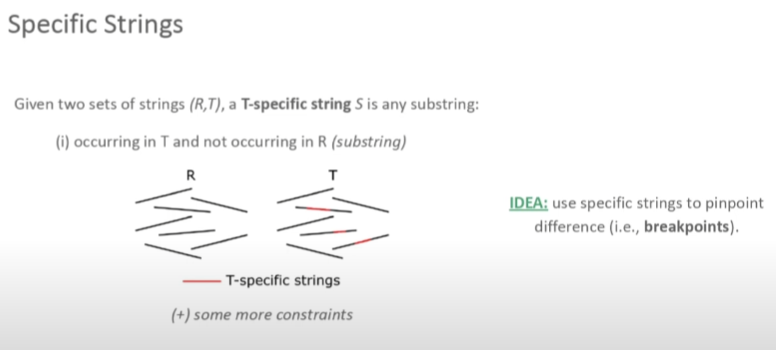
SVDSS融合了基于比对、无比对和基于组装这三种方法的优势。整个流程可以分为如下几步:
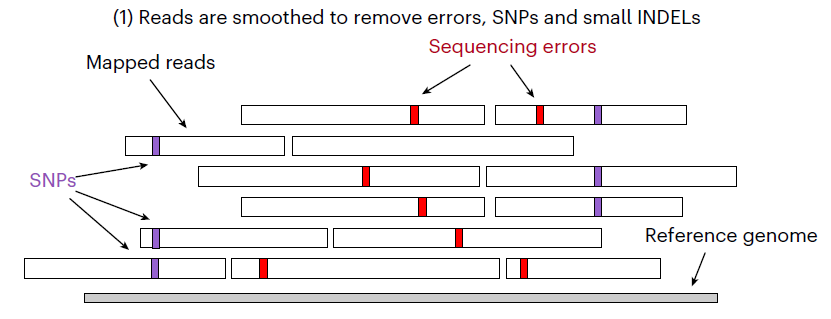
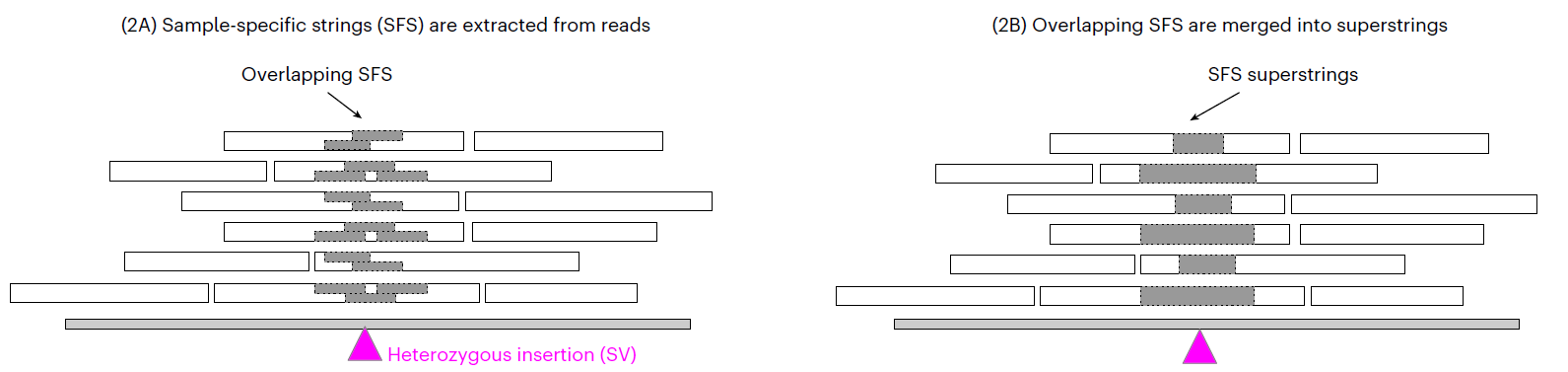
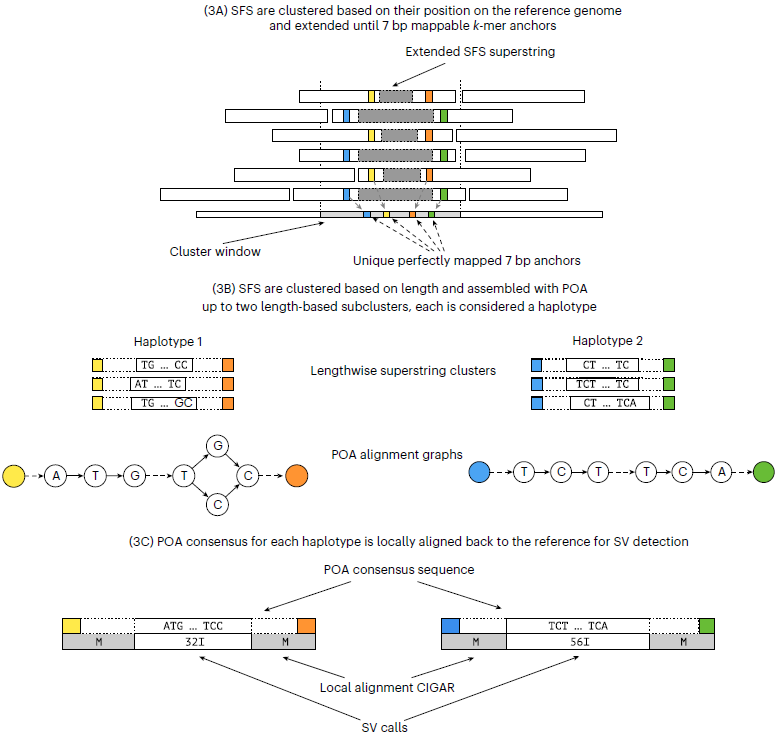
read-smoothing:基于比对到参考基因组上的方法将测序错误碱基、SNP和小片段插入(<20bp)的序列去除。构建SFS superstring:在完成上一步的基础上对样品特异性片段(sample-specific string,SFS)进行提取,然后再将SFS组装成superstrings.SV预测:基于这些SFS在参考基因组上的位置对其进行聚类,然后鉴定SV.
实战案例
## 软件安装
1 2 3 mamba create --name svdss
数据下载
官方使用的是人类基因组数据,我选择水稻的数据。
从[RiceSuperPIRdb ](http://www.ricesuperpir.com/web/download )下载日本晴的T2T基因组和注释信息。从Qin P, Lu H, Du H, et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations[J]. Cell, 2021, 184(13): 3542-3558. e16. 提供的数据中下载2个样品全基因组测序数据。下载地址:
https://ngdc.cncb.ac.cn/search/?dbId=gsa&q=PRJCA002103.&page=1
剩下的步骤大概可以分为如下几步:
Build FMD index of reference genome (SVDSS index)
Smooth the input BAM file (SVDSS smooth)
Extract SFS from smoothed BAM file (SVDSS search)
Assemble SFS into superstrings (SVDSS assemble)
Genotype SVs from the assembled superstrings (SVDSS call)
但是软件输出的帮助文档的顺序和GitHub上的不一样:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 SVDSS, Structural Variant Discovery from Sample-specific Strings.in binary format. Allows for another index to be appended to this index later.for a call to be reported.print version information.help print this help message.
先跟着GitHub上的步骤操作一波试试。
All of SVDSS steps must be run in the same directory so we always pass --workdir $PWD for every command.
构建参考基因组索引
1 2 mkdir 1.refer.index
373M的基因组运行结果如下:
1 2 3 4 5 SVDSS, Structural Variant Discovery from Sample-specific Strings.for this batch as too few strings are left.
Mapping
使用Minimap2将序列比对到参考基因组上获得BAM文件。
1 2 3 minimap2 -ax map-pb ref.fa pacbio-reads.fq > aln.sam
1 minimap2 -t 70 -ax map-pb 00.data/genome/nip.t2t.fa 00.data/sample/zh11.fastq > zh11.sam
57G数据运行了25分钟左右。
将SAM转为BAM排序后构建索引:
1 2 samtools view -@ 70 -b zh11.sam > zh11.bamsort --write-index -@ 70 zh11.bam -o zh11.sorted.bam
Smooth样品
剔除样品中的测序错误碱基、SNP和小片段插入(<20bp)。
1 SVDSS smooth --threads 70 --reference nip.t2t.fa --bam zh11.sorted.bam
构建索引。
1 samtools index -@ 70 -b smoothed.selective.bam
提取样品的SFS并组装
看了下一步的代码里面有一个参数是batches,猜测不同的样本的batches不同,批量处理就不好处理,那就直接一步完成了。
1 SVDSS search --threads 70 --index refer.genome.fmd --bam smoothed.selective.bam
预测SVs
1 SVDSS call --threads 70 --reference nip.t2t.fa --bam smoothed.selective.bam > zh11.sv.vcf
汇总结果
1 2 3 4 5 mkdir 02.each/zh11mv zh11* 02.each/zh11 mv s* 02.each/zh11 mv ignored_reads.txt 02.each/zh11 mv poa.sam 02.each/zh11
另一种方法
我尝试了上面的方法,不行,最终得不到结构变异。选择直接从fastq文件开始,不用比对了。
1 2 3 4 5 6 7 8 9 10 11 12 13 mkdir 1.refer.indexsort --write-index -@ 70 zh11.bam -o zh11.sorted.bam mkdir 02.each/zh11mv zh11* 02.each/zh11 mv s* 02.each/zh11 mv ignored_reads.txt 02.each/zh11 mv poa.sam 02.each/zh11
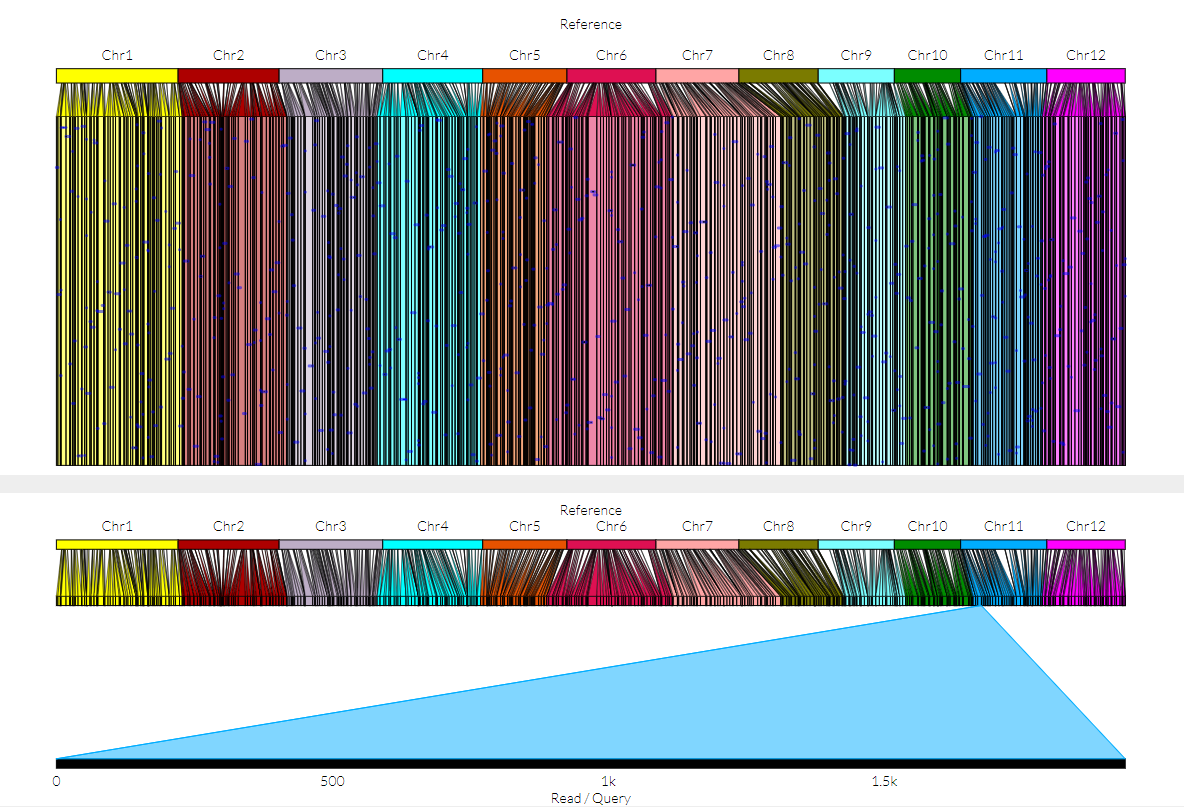
只得到200个SV,不知道对不对。得到这样一个图:
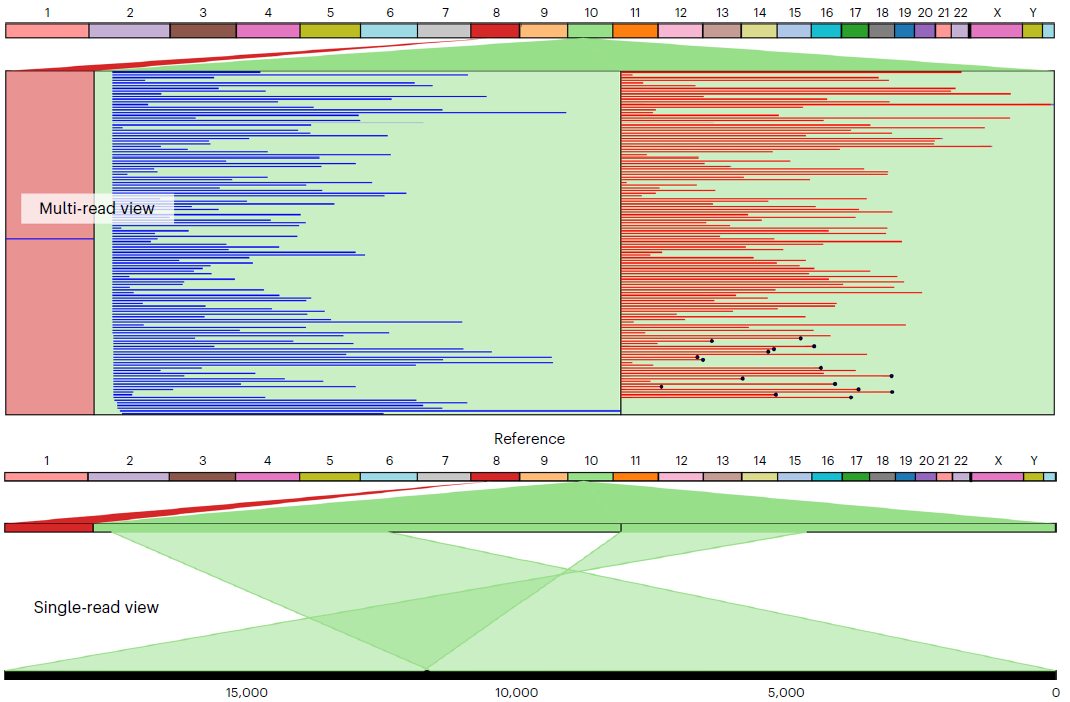
结构变异可视化
在Ahsan M U, Liu Q, Perdomo J E, et al. A survey of algorithms for the detection of genomic structural variants from long-read sequencing data[J]. Nature Methods, 2023, 20(8): 1143-1158. 这个文章里面看到这个图:
找到这个文章:
Maria Nattestad, Robert Aboukhalil, Chen-Shan Chin, Michael C Schatz, Ribbon: intuitive visualization for complex genomic variation, Bioinformatics , Volume 37, Issue 3, February 2021, Pages 413–415
点击查看GitHub代码 。
直接部署一个到自己的服务器上,接个域名方便自己使用。
https://www.xxx.com/ribbon/