R语言笔记
概览

R语言基础
R语言简介
R语言应用
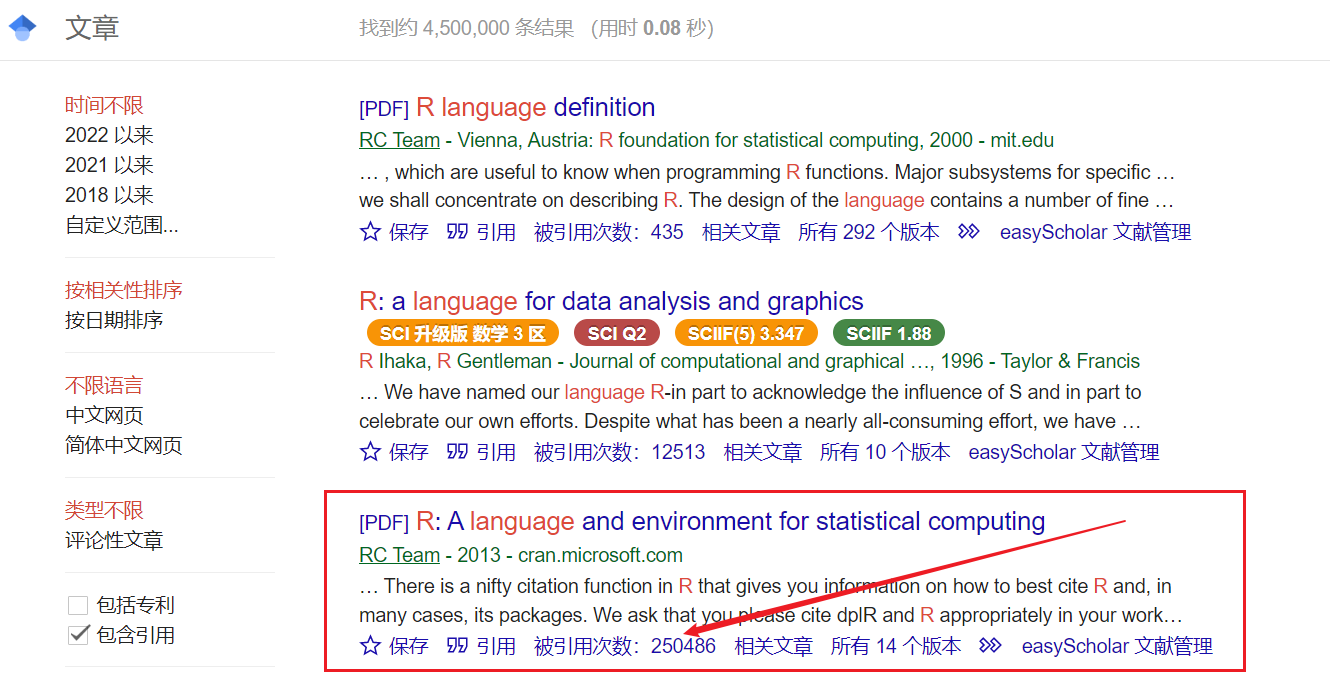
谷歌学术2022年8月10日截图:
软件安装
如果电脑分成系统盘(C盘)和非系统盘(非C盘),先在非C盘的位置创建文件夹R,然后安装R和RStudio的时候选择安装在这个路径。
R
RStudio
rtools
rtools的作用可以简单理解为有时候有些R包的安装需要用rtools编译才能安装。
注意:安装时默认路径安装!!!
软件设置
快捷键
常见报错
R包管理
查找合适的包
R包管理
镜像问题
Bioconductor包
安装R包
R包迁移
文件操作
认识数据
数据清洗
工作目录
读写文本文件
读写EXcel
读写其他文件
数据结构
数据类型
数据结构
变量操作
数据索引
数据索引
判断类型
修改内容
数据操作
合并
排序
筛选
分组
计算
ggplot2基础
资料
绘图语法
- 理解区分分类变量和连续变量;
+必须位于每行绘图代码的末尾;- 区分形状颜色和填充色;

- 不同的几何对象可以筛选数据;
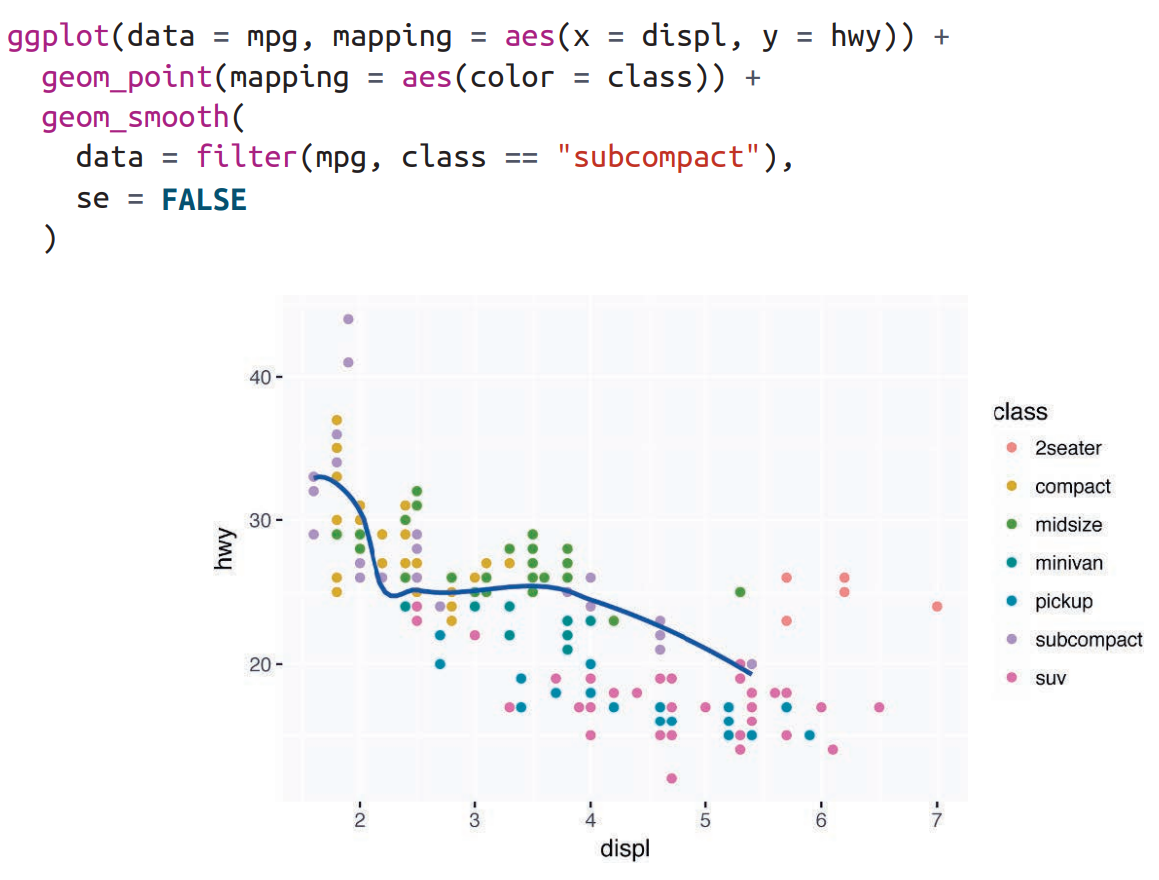
图层概念
主题修改
颜色修改
学习资源
坐标修改
分面操作
常见图形
学习资源
散点图
柱状图
堆积柱状图加上误差线
1 | |
1 | |
直方图
箱线图
韦恩图
小提琴图
热图
基础统计
学习资料
- Summary and Analysis of Extension Program Evaluation in R
- 贝叶斯统计学习
- 现代统计在R中的实现
- 线性回归模型的可视化解释
- R语言是如何实现线性回归模型的
线性模型
求最小二乘均值并标注显著性
1
2
3
4
5
6
7library(tidyverse)
library(multcomp)
library(emmeans)
lm(Sepal.Length ~ Species, data = iris) %>%
emmeans(~Species) %>%
cld(alpha = 0.5, Letters = letters, adjust="tukey")什么是模型的p值:和空模型相比,是否显著。
什么是模型的$R^2$:多少的因变量被模型解释。
glm模型不会返回p值和$R^2$,利用函数anova可以计算p值,函数nagelkerke可以计算伪$R^2$。
t检验
方差分析
卡方检验
wilcox检验
Fisher精确检验
1 | |
贝叶斯统计
学习资料
基础分析
回归分析
相关性分析
机器学习
PCA
LDA
随机森林
1 | |
组学分析
转录组
群体遗传学
GWAS
学习资料
微生物组
大数据挖掘
其他资料
PERMANOVA
1 | |
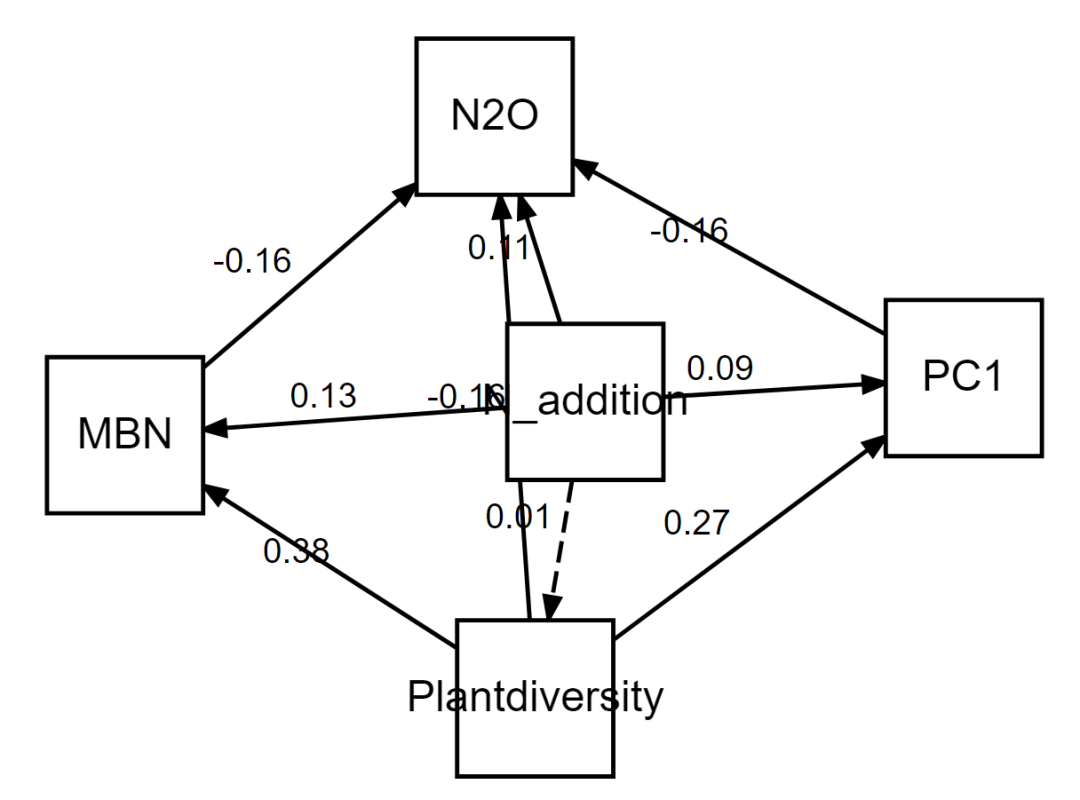
结构方程模型
带线性混合效应模型的 SEM 构建、解释以及实际应用
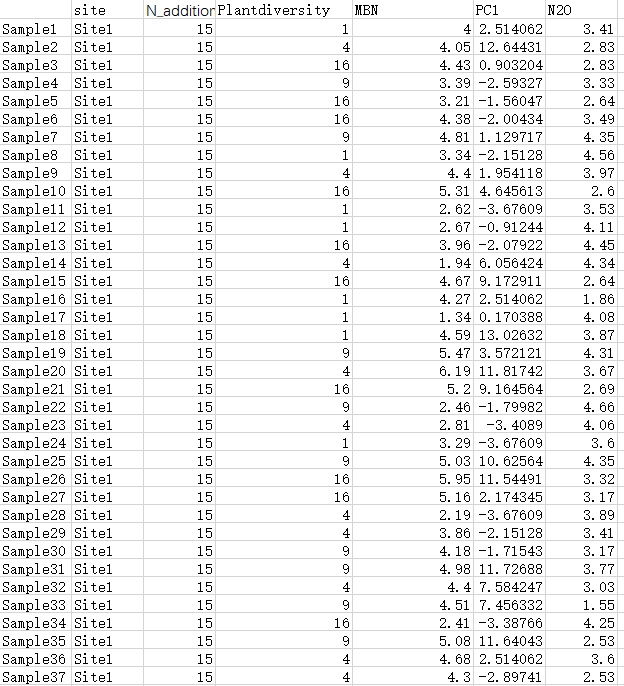
1 | |
R开发
shiny
golem开发shiny
主要笔记:
- 创建项目
下面的代码会创建一个R package格式的文件夹,
1 | |
1 | |
主要的操作都在dev这个文件夹下完成。
- 编辑
DESCRIPTION文件
shinyWidgets
shiny案例
CSS优化shiny
服务器部署Rshiny注意事项
- 应用文件夹应该放在
/srv/shiny-server文件夹下才能生效,否则页面无法找到; - 可以把应用放在
/opt/shiny-server/app/下,然后软连接到/srv/shiny-server.
其他知识点
关于距离
各种距离计算
git知识点
添加服务器git服务器
1 | |
snakemake流程
R语言笔记
https://lixiang117423.github.io/article/sharer/