shiny4qPCR-qPCR数据处理一站式解决方案
为什么做shiny4qPCR
qPCR是现代分子生物学基础,其基本过程涉及引物设计、RNA(DNA)提取、qPCR实验、qPCR数据处理等。从研究生阶段到现在的博士阶段,我需要做大量的qPCR实验,每次数据处理都比较麻烦,虽然已经基本实现流程化了,但还是比较麻烦,因此,以RStudio的shiny-server为基础,写了个shiny app,专门用于处理qPCR相关数据。 注:目前软件源代码尚未开源,因为其中某个算法的部分代码来自其他文献,正在和作者沟通中。
shiny4qPCR 地址:https://shiny.web4xiang.top/shiny4qPCR/

功能模块
shiny4qPCR包含如下几大功能模块:
- 引物设计
- 反转录计算
- 相对标曲计算
- 表达量计算
- 相对标曲法
- 2$^{-\delta\delta C_t}$法
- RqPCR法
- 差异表达统计
下面针对每个模块进行详细使用说明。
引物设计
shiny4qPCR内置的引物设计程序是Primer3$^{[1]}$。目前该软件的引用次数已超过7000次,已经有数十个引物设计软件以该软件为基础$^{[1]}$。该软件在最新版中已经对引物的二级结构、发卡结构等进行优化,而且其还有命令行版本,方便内嵌在shiny server中,因此我们也选择利用{% label Primer3 purple %}作为我们引物设计的底层软件。
参数设置:
- 数据上传
- 单条序列:直接复制粘贴序列即可;
- 多条序列:将序列存在一个
{% label fasta purple %}格式的文件中,上传该文件。
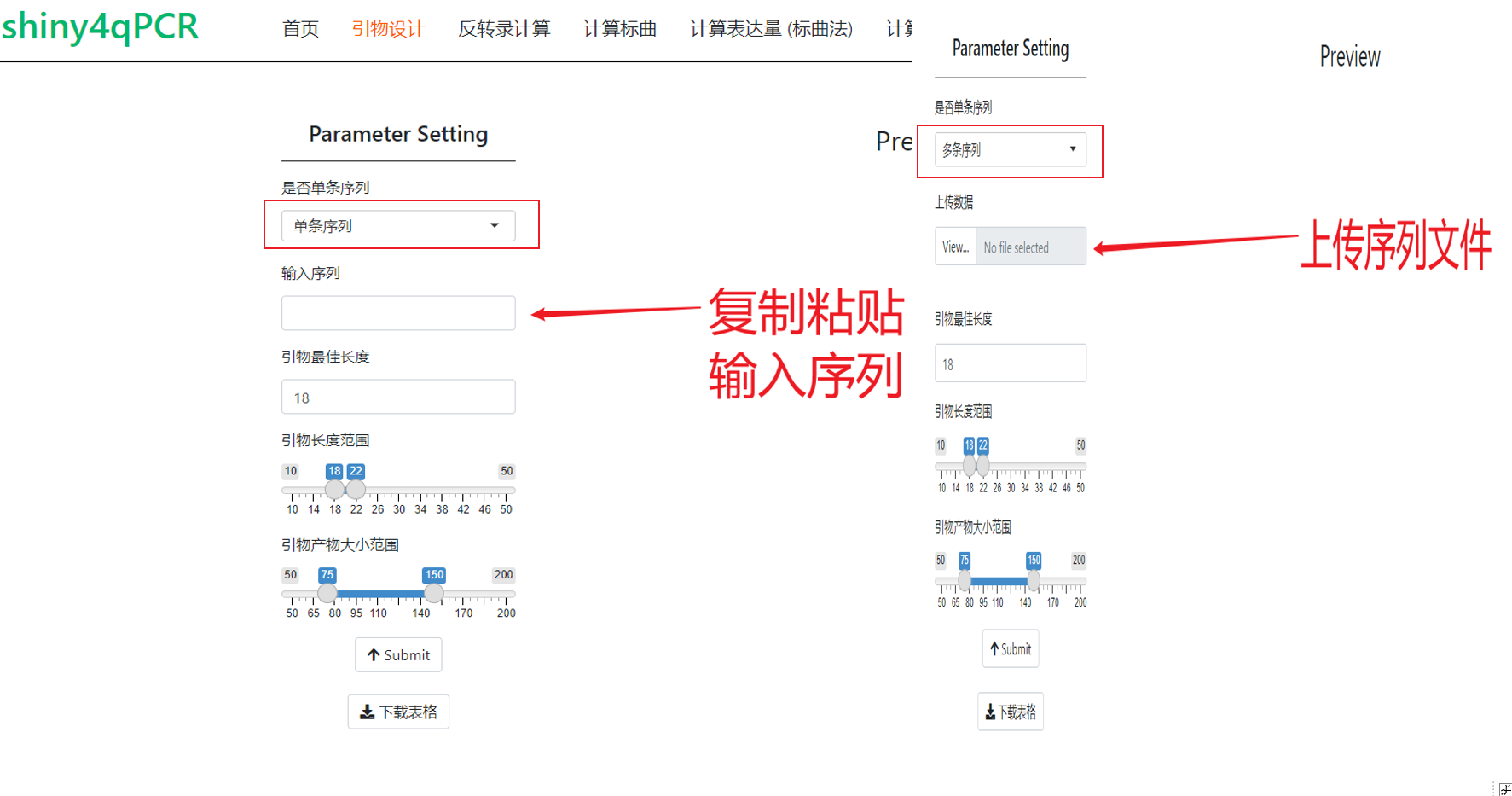
- 引物最佳长度:默认是18
- 引物长度范围:默认18-22
- 引物产物大小范围:默认75-150
参数设置完成后点击{% label Submit purple %}即可,点击{% label 下载表格 purple %}得到的就是输入的序列的所有引物的Excel表。引物适用与否则需要利用PCR进行进一步验证。
反转录计算
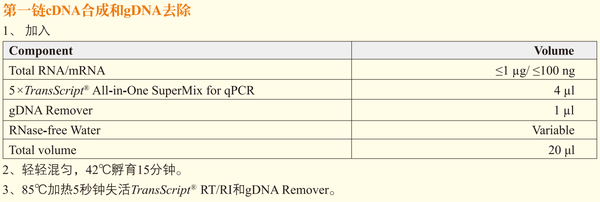
这个功能的应用面比较狭窄,因为是以全式金的反转录试剂盒的说明书(下图)为基础的,目前这个功能只适用于这种试剂盒做反转录的计算。
目前支持的输入数据为NanoDrop 2000的输出数据(下图),我们建议对每个样品至少进行3次平行浓度测定,这样可以保证浓度的稳定性和准确性。如果样品多次测量的浓度差异较大,建议重新涡旋离心后进行测量。
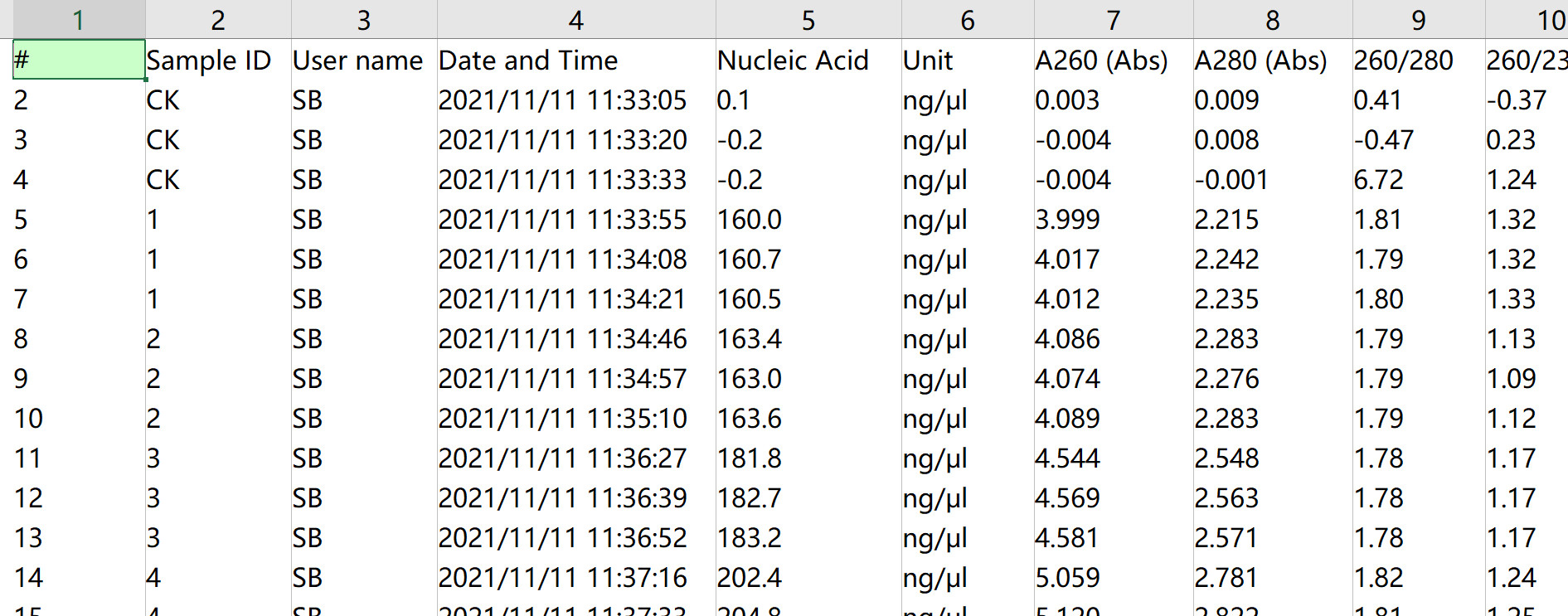
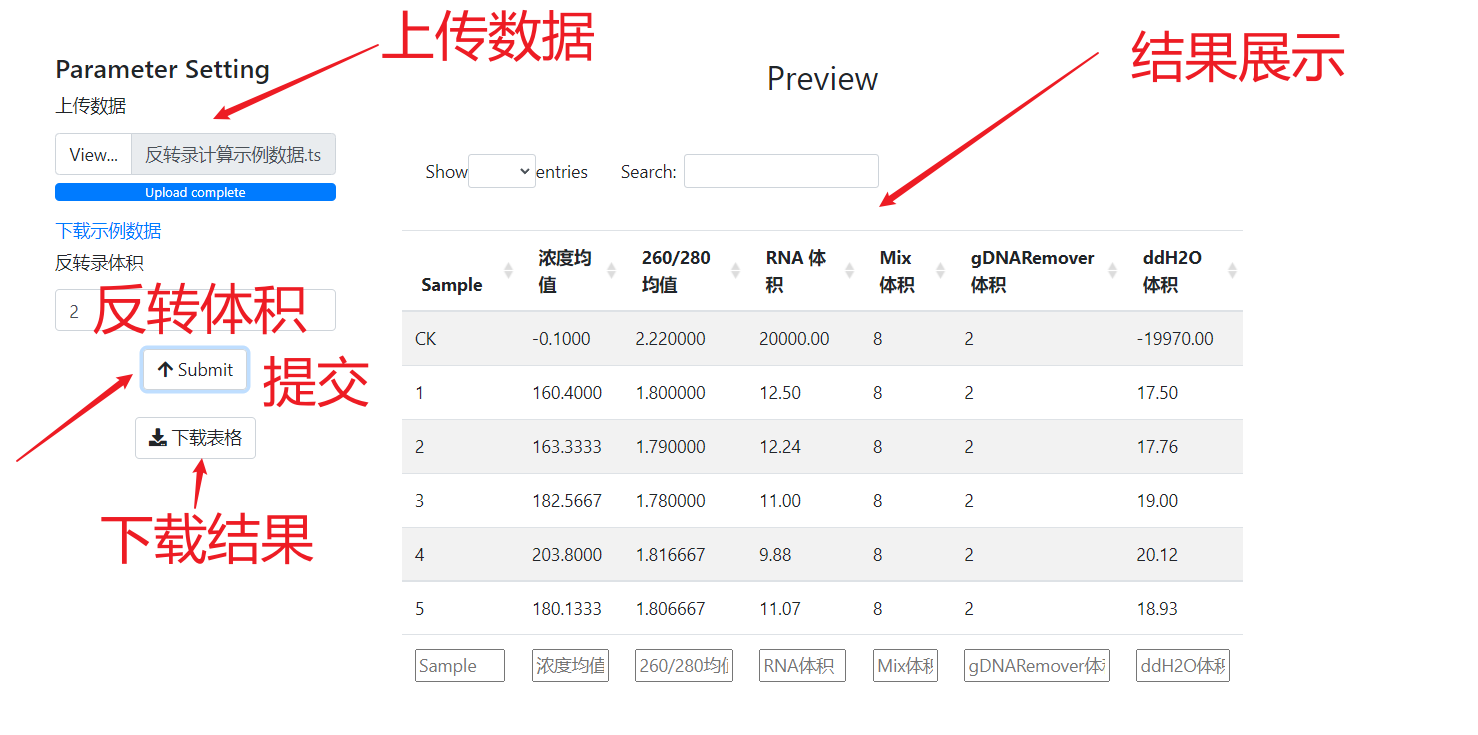
软件当前没有自动删除异常值的功能,因此在上传数据之前需要将每个样品中不需要的值删除(一整行)。
标曲计算
2$^{-\delta \delta C_T}$法是最常用的计算qPCR基因表达量的方法,但是这个方法的一大前提条件是目标基因引物和内参基因引物的扩增效率必须要一致(至少基本一致)。在实际实验过程中是很难达到扩增效率一致的,尤其是当需要检测大量基因时。因此,相对定量就更加适合。该方法不用考虑引物的扩增效率是否一致,每个引物作为独立的扩增对象,对其建立相应的Cq—相对浓度回归曲线,根据曲线去计算某个处理下该基因的相对含量,再进行后续的统计分析。
数据准备
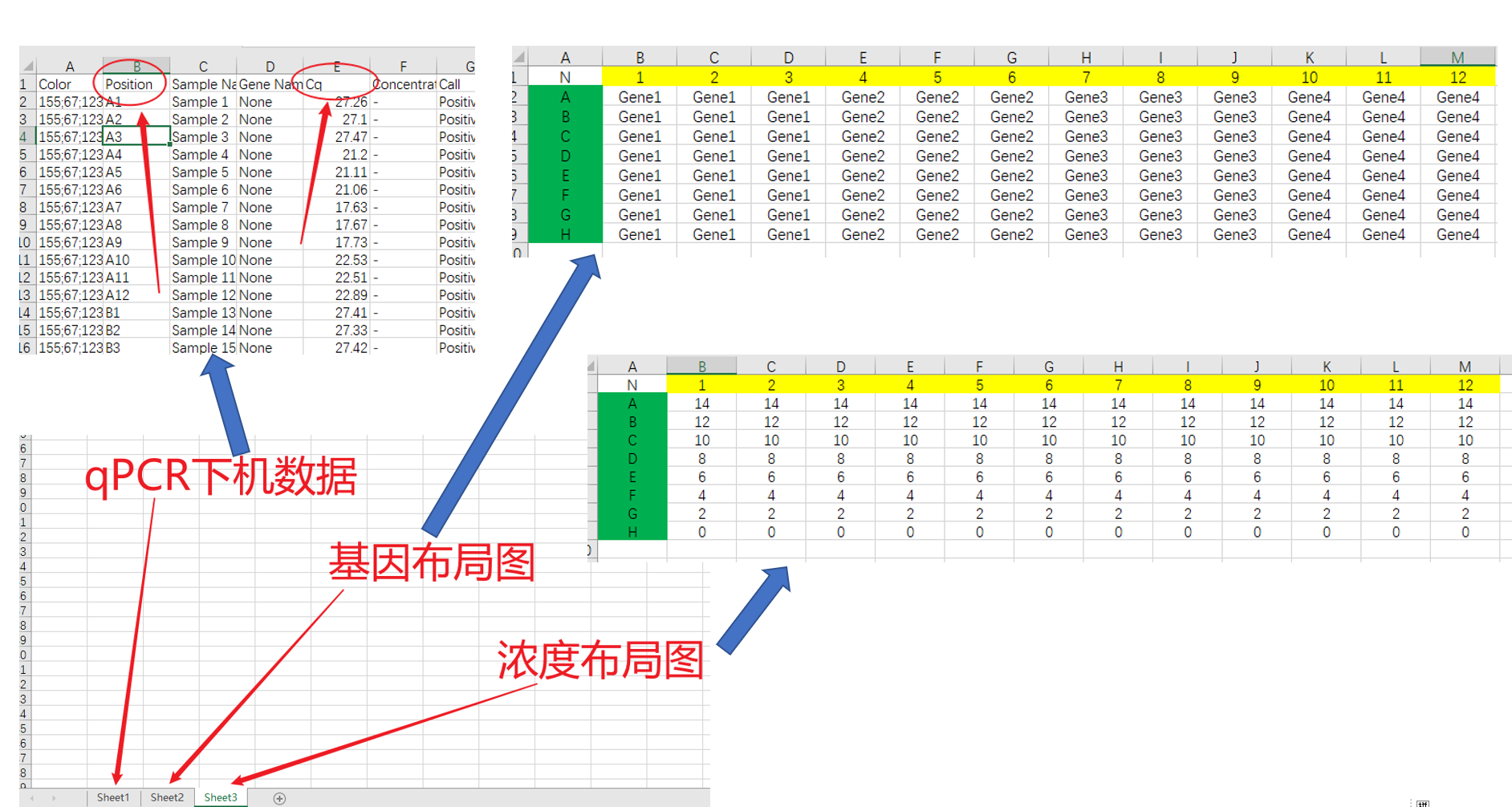
需要准备三个表格(下图):
- qPCR下机数据:最重要的是
{% label Position purple %}和{% label Cq purple %}这两列,软件会自动识别提取这两列的内容进行后续的计算。 - 基因布局图:简单来说就是96孔板的每个孔里面分别是那个基因,这个需要严格区分大小写,因为后续进行表达量计算的时候是根据基因进行匹配的。
- 浓度布局图:简单来说就是96孔板的每个孔里面底物的浓度(相对浓度)。稀释倍数要严格一致,最低浓度为1,倒数第二的浓度应该就是1×稀释倍数,以此类推。如果系数倍数是4,那浓度从低到高应该是1,4,46,64,256 ……;如果稀释倍数是10,那浓度从低到高应该是1,10,100,1000,10000 ……
参数设置
- 上传数据:上传上一步准备好的数据即可;
- 是否剔除空值:在qPCR检测的过程中很多原因会导致某个孔无法进行扩增,在导出结果的时候其Cq值就是
{% label - purple %}。这个时候就需要对这种值进行剔除。软件默认的是剔除这类值,如果不进行剔除,软件无法进行计算。 - 空值填充方法:软件考虑到完全剔除空值后对应的重复数量就减少了,因此选择对空值进行填充,默认的填充方式是均值填充,也就是利用某个基因在某个浓度下的非空值的均值去填充该基因在该浓度下的空值。
- 最低浓度:选择用于标曲计算的最高浓度,默认的是
{% label 4096 purple %}(4倍稀释时候的第二个最高的浓度)。 - 最高浓度:选择用于标曲计算的最低浓度,默认的是
{% label 4 purple %}(4倍稀释时候的倒数第二个最高的浓度)。 - 图片格式:程序默认绘制对应的回归曲线图,点击
{% label 下载图片 purple %}即可进行下载。
数据上传完成、参数设置完成后点击{% label Submit purple %}便开始进行计算。回归方程相关信息展示在右面,点击{% label 下载表格 purple %}即可下载Excel格式的回归方程结果。
注意事项
返回的结果中的
{% label Eff purple %}指的是该引物的扩增效率,正常来说引物的扩增效率的范围是[0,1]之间,结果中的引物扩增效率是计算的引物的扩增效率+1后的结果,这样做是为了后续计算表达量取对数的时候得到无穷大值。上传的数据必须是Excel格式的数据,三个表格的顺序不能错;
- 基因布局图和浓度布局图必须要有A-H和1-12以及左上角的
{% label N purple %}。
引物扩增效率公式推导
引物质量是{% label qPCR purple %}实验中至关重要的一步,网上的教程大多是以10倍稀释为例子讲解如何求引物的扩增效率。但是在实际实验中,不一定都是10倍稀释的。因此,我就把引物扩增效率的通用计算公式推导了一遍。
PCR扩增的通用公式:
其中$X_n$是最终的拷贝数,$X_0$是起始拷贝数,$E_x$是该基因引物的扩增效率,$n$是循环数。理想状态下扩增效率为1。
现在假设我们有如下浓度梯度及其$C_q$值:
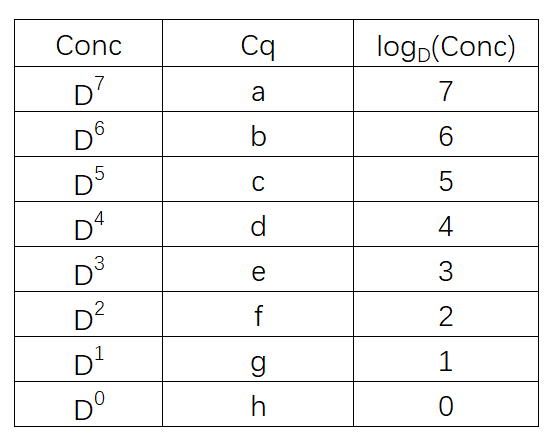
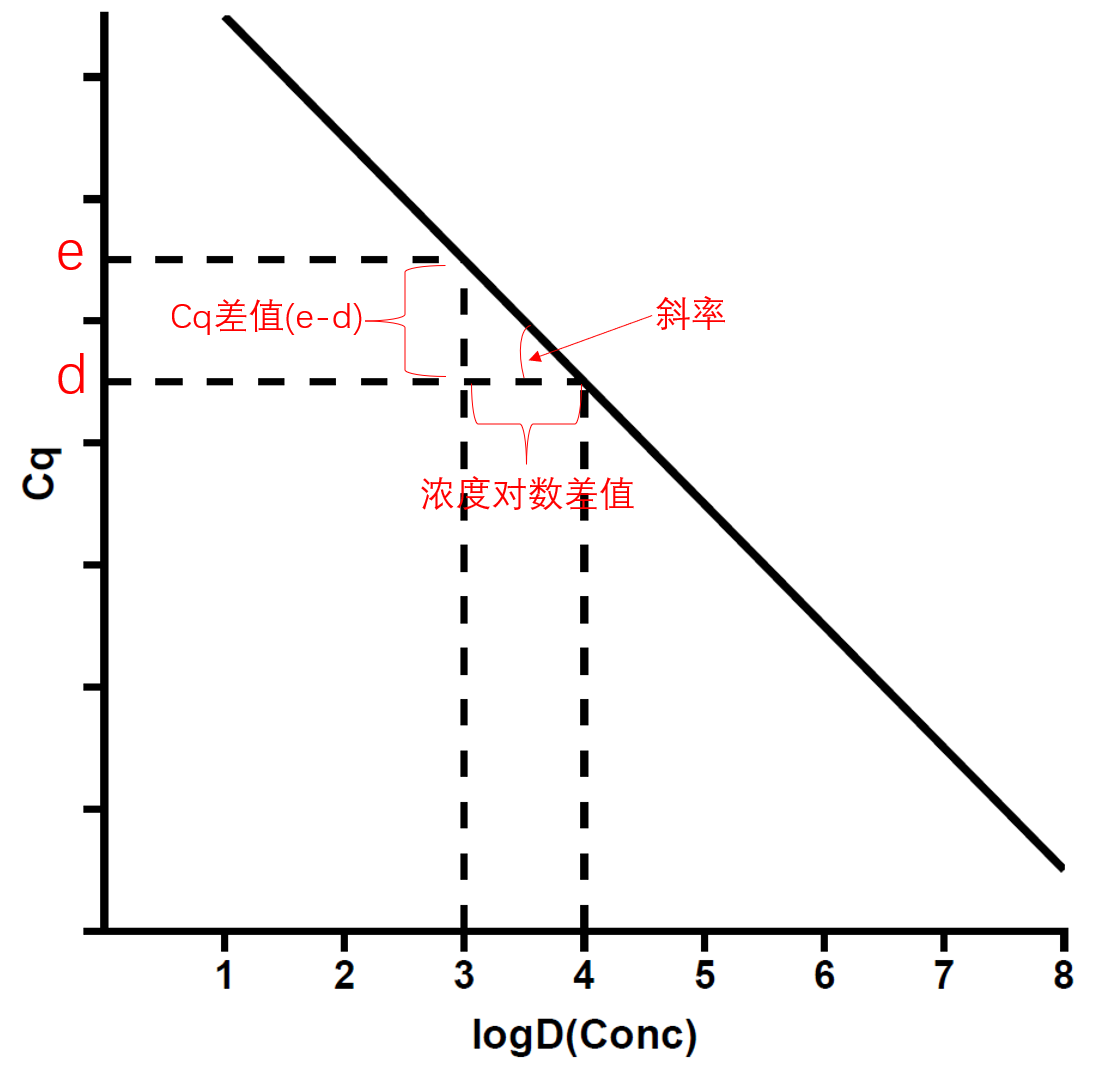
然后我们用图画出来:
假设现在某个基因我需要扩增到$X_n$个拷贝,我现在有两个浓度的模板cDNA,分别是$D^4$和$D^3$。则有:
也就是分别经过d次和e次扩增后,最终达到$X_n$个拷贝,那么也就有:
等式两边交叉相除:
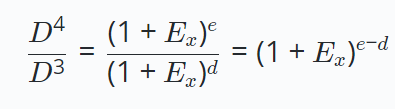
那么,最左边相除以后得到的{% label D purple %}刚好是稀释倍数的D,右边的$e-d$刚好是两条扩增曲线的$C_q$值的差值。
那么当引物的扩增效率的等于1的时候:
当稀释倍数为10时:
可以计算出$C_q差值 = 3.321928$。也就是10倍稀释的时候相邻两条扩增曲线之间的$C_q$值的差值应该是3.321928。
现在我们假设稀释倍数为$D$,也就有:
从上面的图我们可以看到的是,$e - d$ 刚好就是这条直线的斜率(slope)$\frac {d-e}{4-3} = d-e$ 的相反数,那就有:
等号两边同时以$D$为底数取对数:
所以:
也就是当知道稀释倍数和斜率的时候就可以计算引物的扩增效率了。
表达量计算
标曲法
数据准备
需要4个数据,4个数据存放在一个Excel文件种:
- qPCR下机数据:比较重要的三列数据:
{% label Batch purple %}:可以理解成第几板qPCR,因为在实际操作过程中很可能不止有一板qPCR,有很多板的情况下加上{% label Batch purple %}这个参数就能一次完成计算。{% label Position purple %}:告诉程序分别是哪个孔。{% label Cq purple %}:每个孔对应的Cq值。
- 处理布局图:每个孔内的样品来自什么处理。
- 基因布局图:每个孔内的基因叫什么。
- 标曲:前一步计算的标曲(不能做任何修改,直接复制粘贴)。

参数设置
比较重要的两个参数:
- 是否用内参进行校正:标曲法计算得到的相对向量通常需要用内参基因的表达量进行校正,程序默认使用内参进行校正。
- 内参基因名称:输入的基因名称必须和基因布局图以及标曲表对应起来。
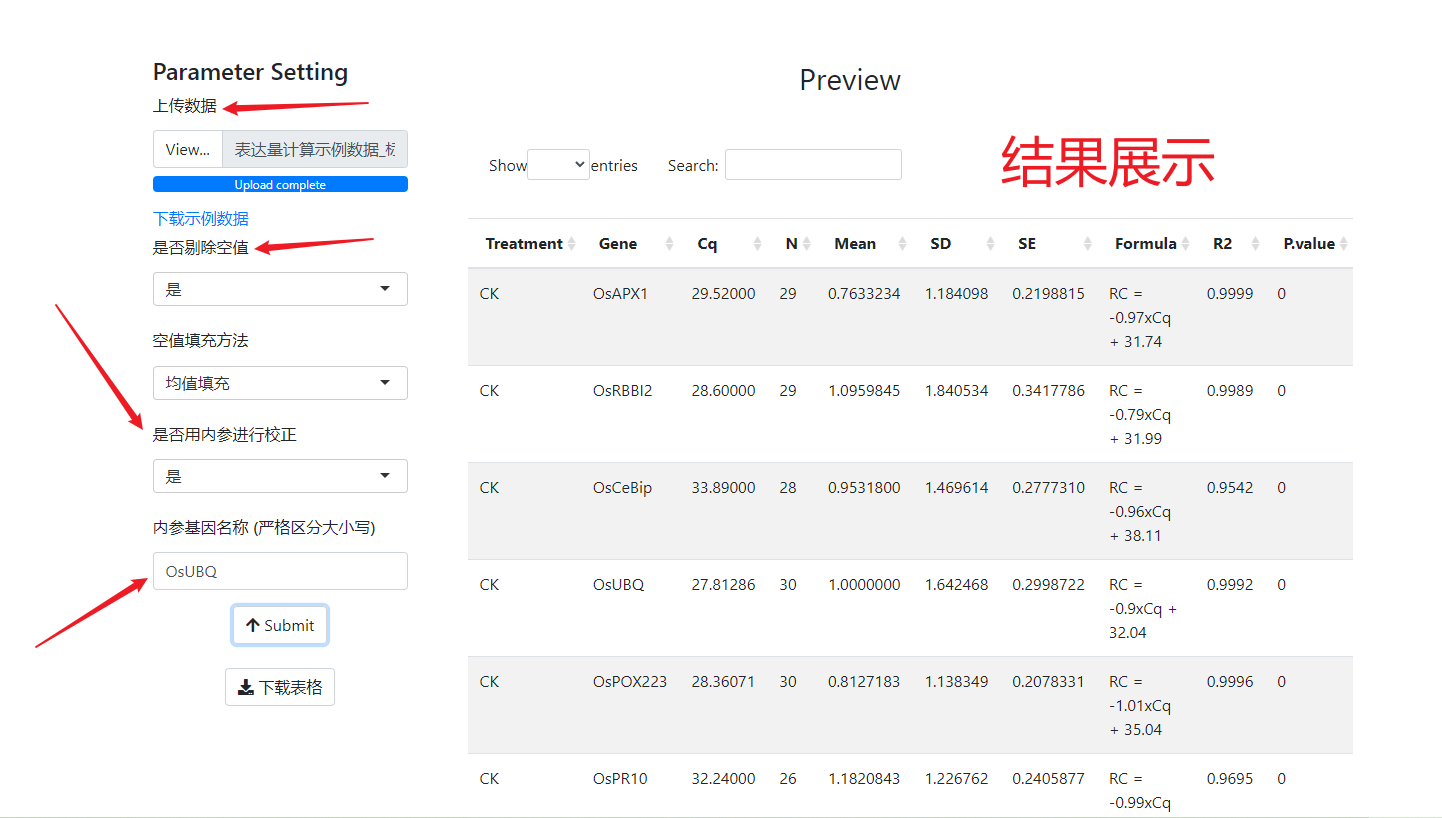
结果解读
下载得到的表格是个Excel表,含有两个Sheet:
- 第一个Sheet是均值表达量,也就是对不同处理的不同基因的表达量求均值后输出;
- 第二个Sheet是每个孔的表达量,也就是输入的数据有多少个孔,返回的数据与其一一对应。
注意事项
- 4个文件必须放在一个Excel文件中,且顺序不能乱;
- 前三个表必须含有
{% label Batch purple %}这一列,哪怕是只有1板qPCR,也得把{% label Batch purple %}这一列加上; - 基因布局表和标曲表的基因名称必须完全对应,严格区分大小写。
RqPCR法
{% label RqPCR法 purple %}是根据Frédérique Hilliou等$^{[2,3]}$的算法进行开发的。
注:如果您使用这种方法计算表达量,请引用他们的文章:
- Rancurel C, Van Tran T, Elie C, et al. SATQPCR: Website for statistical analysis of real-time quantitative PCR data[J]. Molecular and cellular probes, 2019, 46: 101418.
- Hilliou F, Tran T. RqPCRAnalysis: Analysis of Quantitative Real-time PCR Data[C]//Bioinformatics. 2013: 202-211.
算法特点
该方法的特点在于不用提供参考基因,而是由软件计算完后自动寻找该批次数据中的参考基因。该算法默认至少选择两个基因作为参考基因,因此至少需要检测3+的基因才能用这种方法。
数据准备
由于该算法以根据不同处理的不同生物学重复进行分批计算的,因此共需要6个数据,将6个数据按顺序存放在一个Excel文件中。下载示例数据讲对应的数据替换成自己的数据即可。
- 下机数据:主要包含3行信息:
- Batch:第几批次(第几版qPCR下机数据)
- Position:对应哪个孔。
- Cq:Cq值。
- 处理布局:每个孔对应的是哪个处理。
- 基因布局:每个孔对应的是哪个基因。
- 生物学重复布局:每个孔对应的是哪个生物学重复。
- 技术重复布局:每个孔对应的是哪个技术重复。
- 引物扩增效率:每个基因的引物的扩增效率。此处的扩增效率是
{% label 扩增效率+1 purple %}的结果。也就是前一步计算标曲的时候得到的扩增效率。
参数设置
- 是否指定参考基因:默认不指定参考基因,程序自动计算参考基因,默认是2个参考基因;如果指定参考基因,那程序就会以输入的参考基因作为计算基础;
- 是否用某个样品校正表达量:程序默认的是用同一个基因下不同处理间最小的表达量作为校正因子校正该基因在不同处理中的表达量;如果指定了用于校正的样品,那么就会以该样品的表达量去校正其他样品的表达量。
注意事项
- 该算法推荐程序自动计算参考基因,默认的参考基因数量是2个,不能少于2个;如果指定参考基因,也可以,不过不推荐。
- 基因的扩增效率需要提前计算好,而且第六个表
{% label 引物扩增效率 purple %}里面的扩增效率必须是{% label 1+引物扩增效率 purple %}。如果没有测定扩增效率,就全部写为2,默认扩增效率为1。扩增效率大小会影响最后的表达量。 - 程序默认最后的表达量是用所有处理中最小的表达量进行校正的,如果选择用某个处理进行校正,就会以该处理的表达量对其他表达量进行校正。
- 进行
{% label t-检验 purple %}的时候将结果中的{% label Exore4Stat purple %}取对数{% label log2() purple %}后再进行统计分析。
2(-🔺🔺Ct )法
qPCR表达量计算使用最广泛的方法毫无疑问是$2^{-\delta \delta C_t}$法$^{[4]}$。但是该方法的前提条件是目的基因和参考基因的扩增效率必须一致(接近1)才行,文章里面多次提到这个关键之处:

为什么要扩增效率一致才能这样计算呢?根据前面我们推导的扩增效率的可知:
那么就有:
当稀释倍数一致的时候,扩增效率的大小严重影响$C_q$值,很小的$C_q$值差异在经过2的次幂以后,会变得较大,有可能夸大了生物学处理之间的真正差异。因此,在使用这个方法之前,最好是先跑个标曲看看引物的扩增效率是否一致且接近于1。
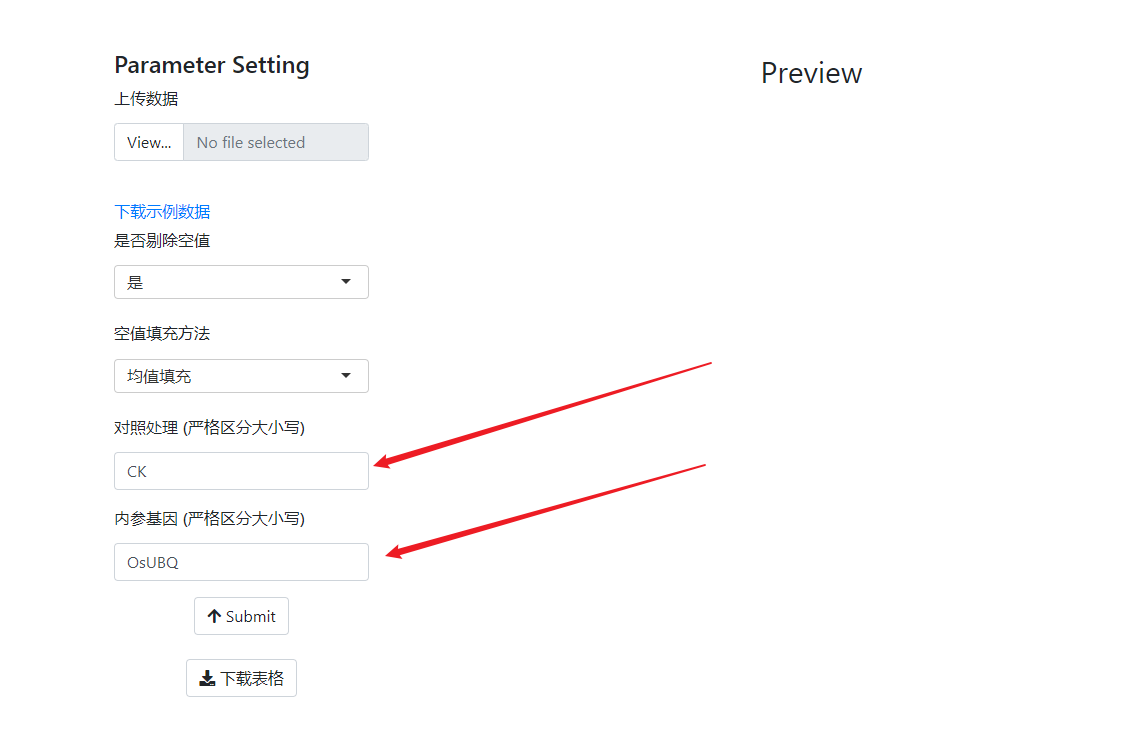
数据准备
数据准备和标曲法是一致的,唯一的差别在于不需要标曲文件。
参数设置
由于2$^{-ΔΔC_t}$法需要对照处理(CK)和内参基因,因此需要在参数设置时指定对照处理的名称和内参基因的名称,注意严格区分大小写。
差异表达统计
数据准备
默认使用的数据类型是前面计算表达量得到的结果,主要是三列:
{% label Treatment purple %}:处理名称{% label Gene purple %}:基因名称{% label Expression purple %}:表达量
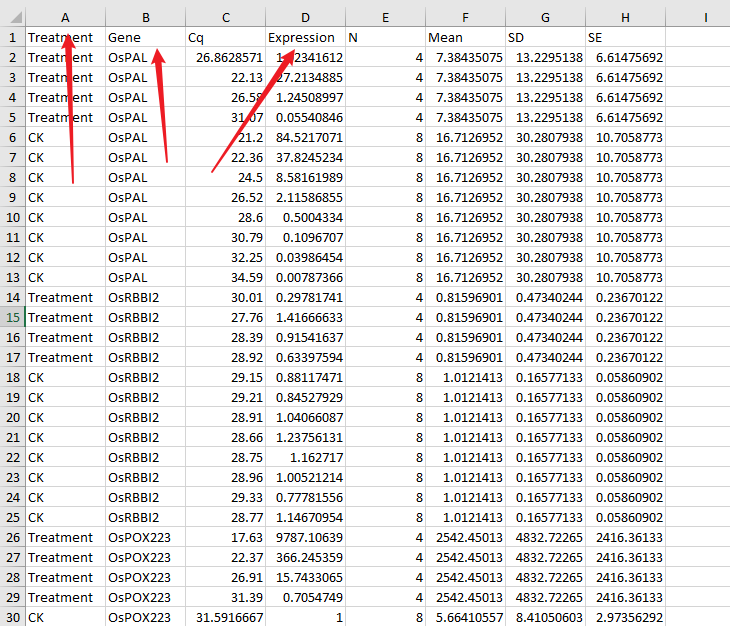
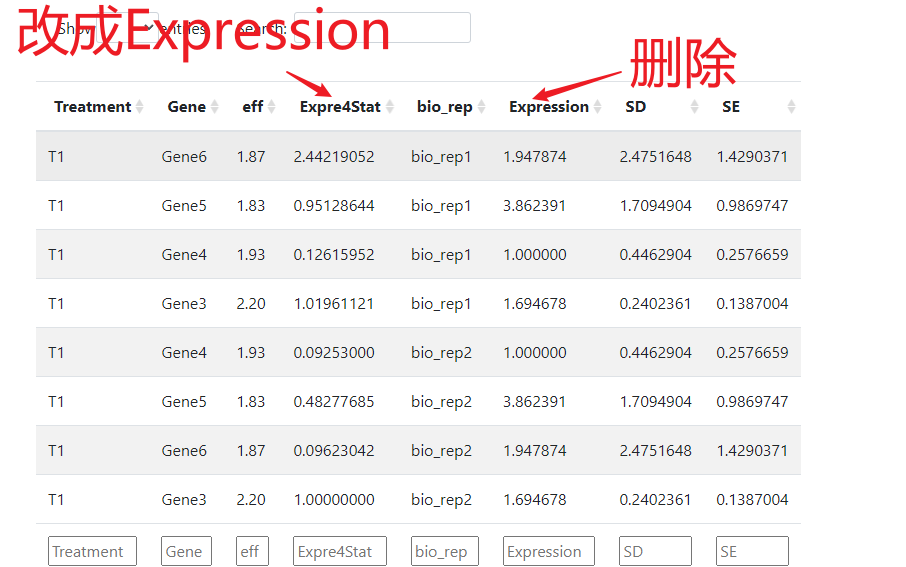
注意:如果表达量计算使用的方法是{% label RqPCR法 purple %}的话,需要将{% label ExpressionStat purple %}改成{% label Expression purple %}才行,而且需要将原来的{% label Expression purple %}删掉才行(或者重命名)。
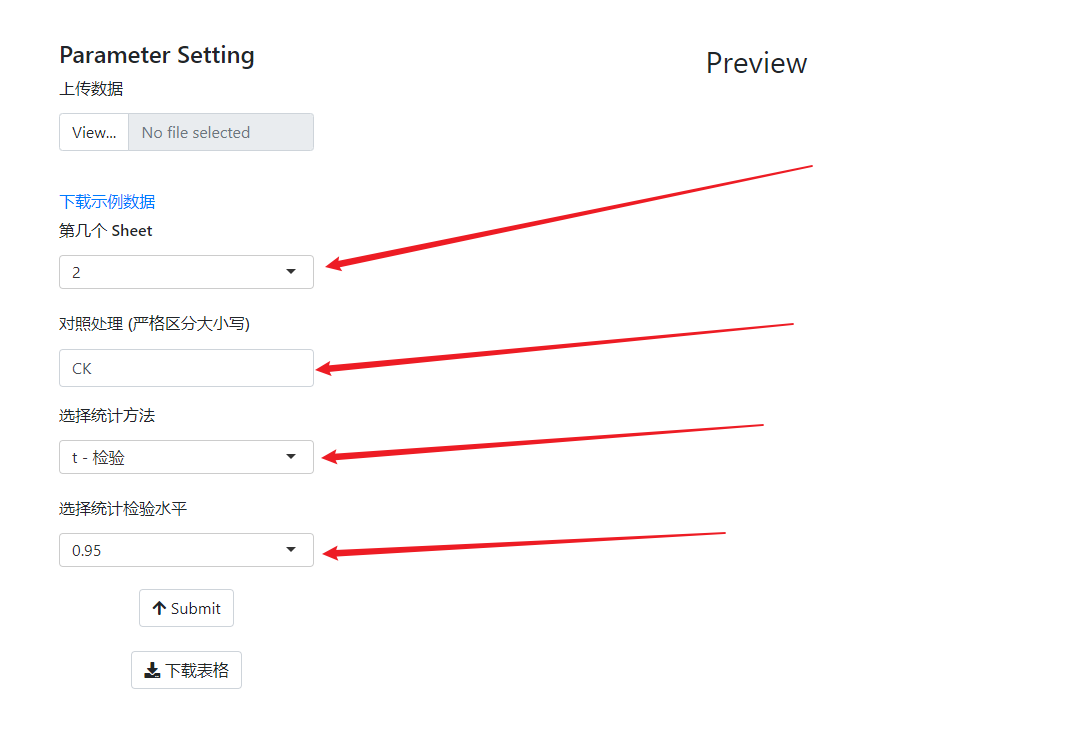
参数设置
{% label 第几个Sheet purple %}:由于前面计算表达量输出的结果的第二个Sheet才是原始表达量,因此程序默认表达量是在第二个Sheet,可以根据自己的数据进行修改。如果是自己的数据,那改成对应的Sheet即可。{% label 统计检验方法 purple %}:目前程序内置了t-test和Anova(with Tukey),如果只是有一个处理+一个对照,那推荐选择{% label t-test purple %},多个处理时推荐使用{% label Anova purple %}。{% label 统计检验水平 purple %}:默认统计检验水平为0.95,可以自行设置。
参考文献
💌lixiang117423@foxmail.com
💌lixiang117423@gmail.com