QIIME2学习笔记
安装
直接用Docker安装。
官方教程:https://docs.docker.com/engine/install/ubuntu/
数据
QIIME2中的数据:
- 测序数据:
.qza格式。因此需要先将测序下机的.fasta格式的文件转换成.qza格式的文件。同样,.qza格式文件也可以导出成其他格式。 - 样品信息(metadata)
- 可视化数据:
.qzv格式。这个文件是分析过程中产生的临时文件。这中类型的数据是不能用作其他分析的输入数据的 。
QIIME工作流程
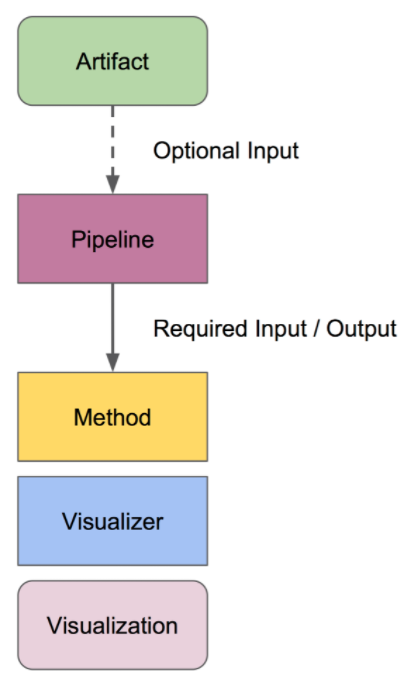

- 拆分
为什么要拆分样品呢?现代的测序仪在一个lane/run中能够对成百上千个样品进行测序,因此需要按照样品将它们分开。如何区分不同的样品呢?用barcode进行区分,也就是接头序列。q2-demux或者q2-cutadapt。如果序列中含有引物序列的时候用第二个程序。
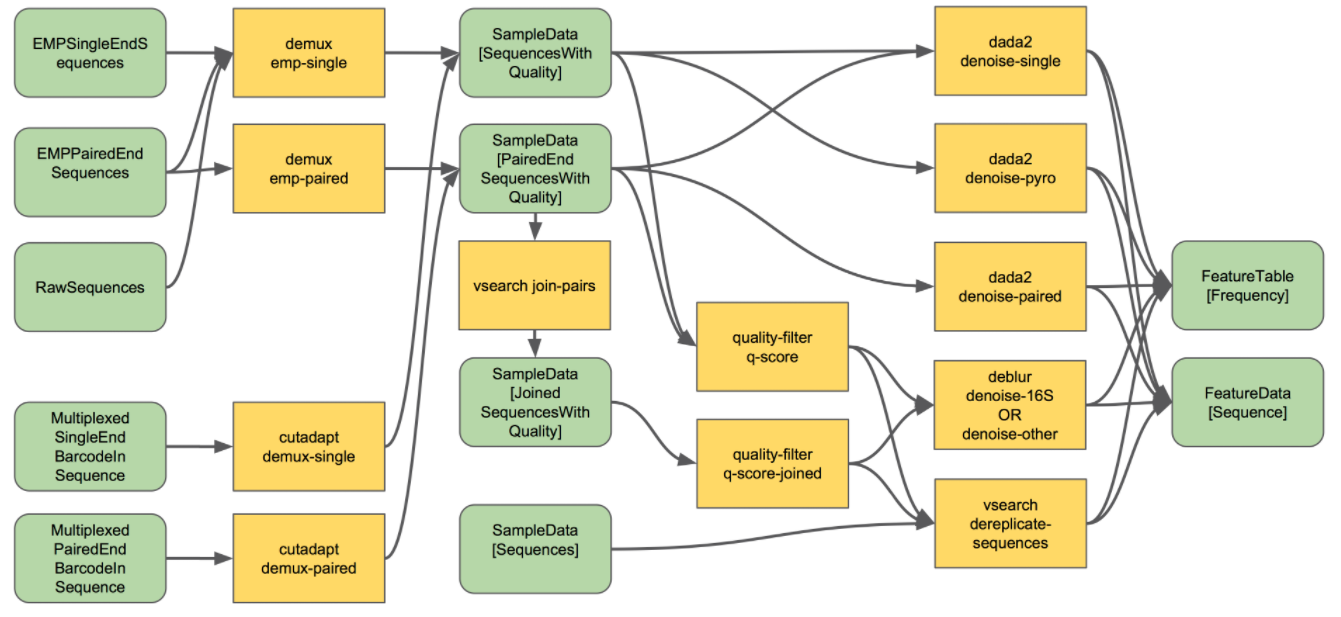
去噪
- DADA2
- Deblur
聚类
classify-consensus-blastandclassify-consensus-vsearchare both alignment-based methods that find a consensus assignment across N top hits.如果用机器学习进行分类的话,需要自行训练参考基因集。
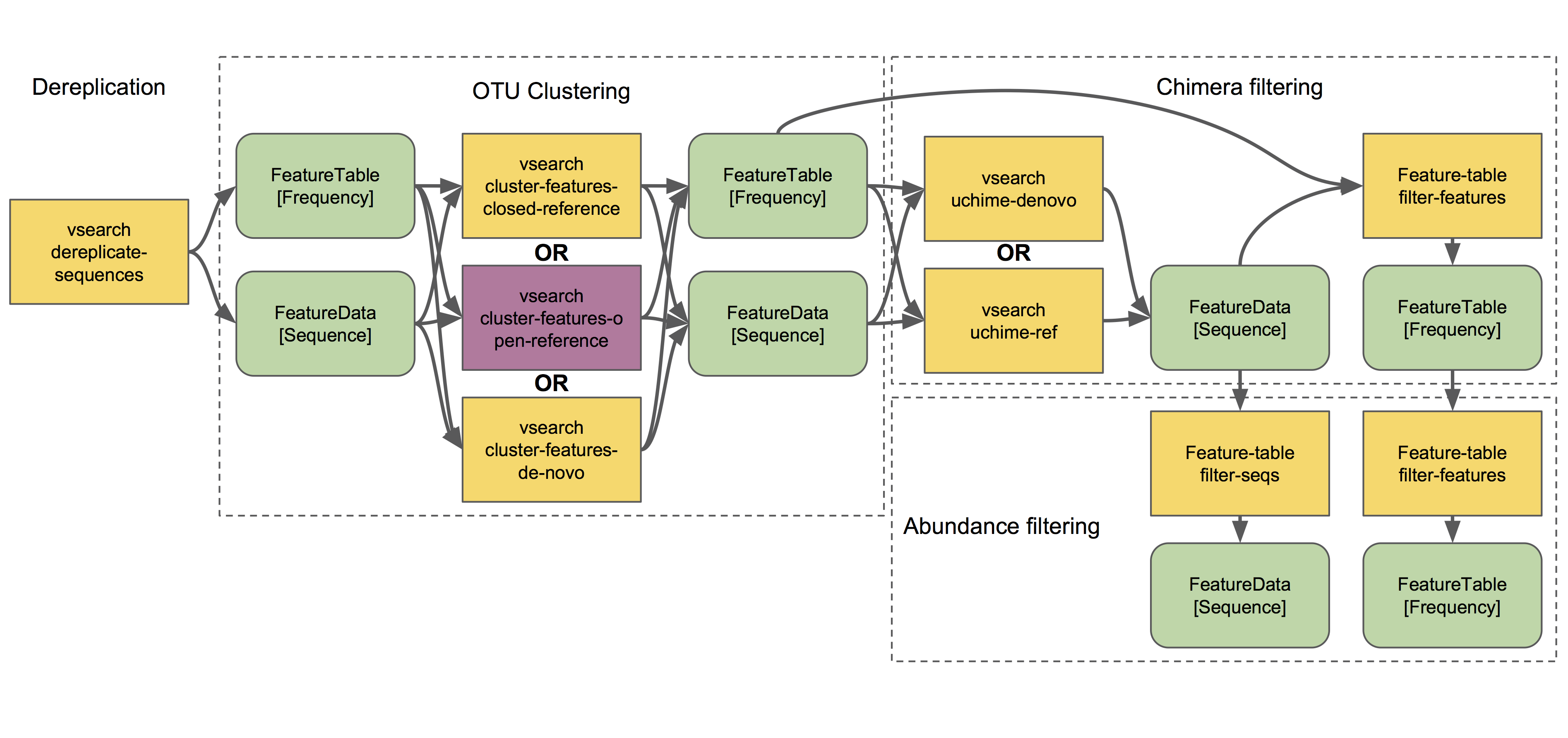
ASV或OTU表
- 降低测序错误
- 去除重复序列
特征表:样品x观测值。几乎参与了所有的分析步骤。
- 序列分类,门纲目科属种

- $\alpha$多样性(样品内)和$\beta$多样性分析(样品间)
- 以系统发育相似度为基础的其他多样性分析,如16sRNA基因为基础的系统发育关系分析
- 差异丰度分析
代表性序列
单末端数据分析案例
教程地址:https://docs.qiime2.org/2021.11/tutorials/atacama-soils/
- 数据下载
将barcode和sequences序列放在同一个文件夹下。
- import数据
1 | |
metadata不能和这两个序列文件放在一起,不然会报错。
- 检查数据的类型等
1 | |
1 | |
- 按样品拆分序列
1 | |
- 查看序列拆分情况
1 | |
生成的文件需要浏览器才能打开。
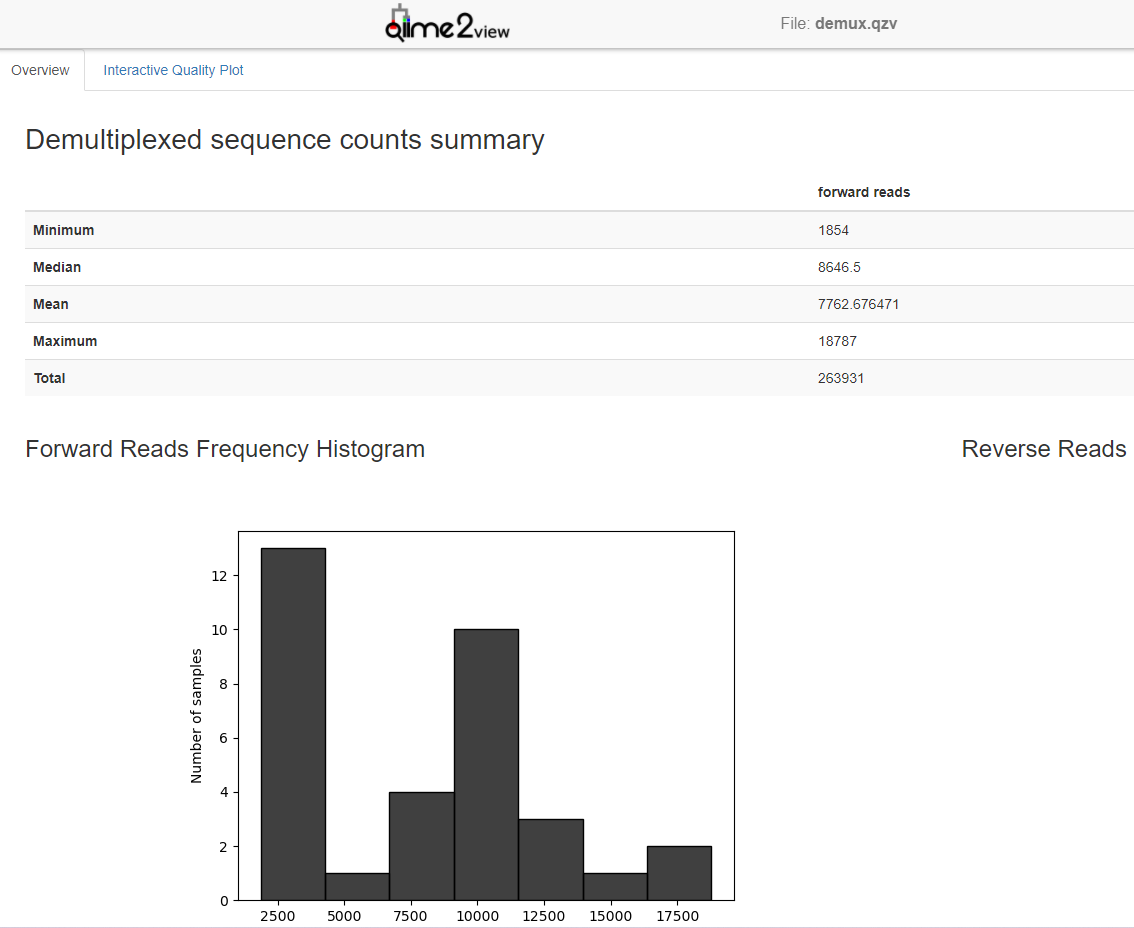

序列质控
DADA2:两个参数
--p-trunc-len n在位置n的地方截断序列--p-trim-left m切除最开始的m个碱基
从上面的图可以看出的是,起始序列的质量是很OK的,在120bp位置左右,序列的质量出现明显的下降。因此就不对起始序列进行裁剪,然后在120bp位置进行裁剪。
1
2
3
4
5
6
7
8
9
10
11qiime dada2 denoise-single \
--i-demultiplexed-seqs step2_demux/demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences step3_dada2/rep-seqs-dada2.qza \
--o-table step3_dada2/table-dada2.qza \
--o-denoising-stats step3_dada2/stats-dada2.qza
qiime metadata tabulate \
--m-input-file step3_dada2/stats-dada2.qza \
--o-visualization step3_dada2/stats-dada2.qzv生成的
.qzv文件直接拖入到QIIME2官网即可查看。Deblur
质量控制
1
qiime quality-filter q-score --i-demux step2_demux/demux.qza --o-filtered-sequences step4_deblur/demux-filtered.qza --o-filter-stats step4_deblur/demux-filter-stats.qza正式程序
1
2
3
4
5
6
7/data# qiime deblur denoise-16S \
> --i-demultiplexed-seqs step4_deblur/demux-filtered.qza \
> --p-trim-length 120 \
> --o-representative-sequences step4_deblur/rep-seqs-deblus.qza \
> --o-table step4_deblur/table-deblur.qza \
> --p-sample-stats \
> --o-stats step4_deblur/deblur-stats.qza可视化结果
1
2
3
4
5
6
7qiime metadata tabulate \
--m-input-file step4_deblur/demux-filter-stats.qza \
--o-visualization step4_deblur/demux-filter-stats.qzv
qiime deblur visualize-stats \
--i-deblur-stats step4_deblur/deblur-stats.qza \
--o-visualization step4_deblur/deblur-stats.qzv
特征表和特征序列
DADA2
1
2
3
4qiime feature-table summarize \
--i-table step3_dada2/table-dada2.qza \
--o-visualization step5_feature_counts_and_seqs/table_dada2.qzv \
--m-sample-metadata-file sample-metadata.tsv
1
2
3qiime feature-table tabulate-seqs \
--i-data step3_dada2/rep-seqs-dada2.qza \
--o-visualization step5_feature_counts_and_seqs/rep-seqs-dada2.qzv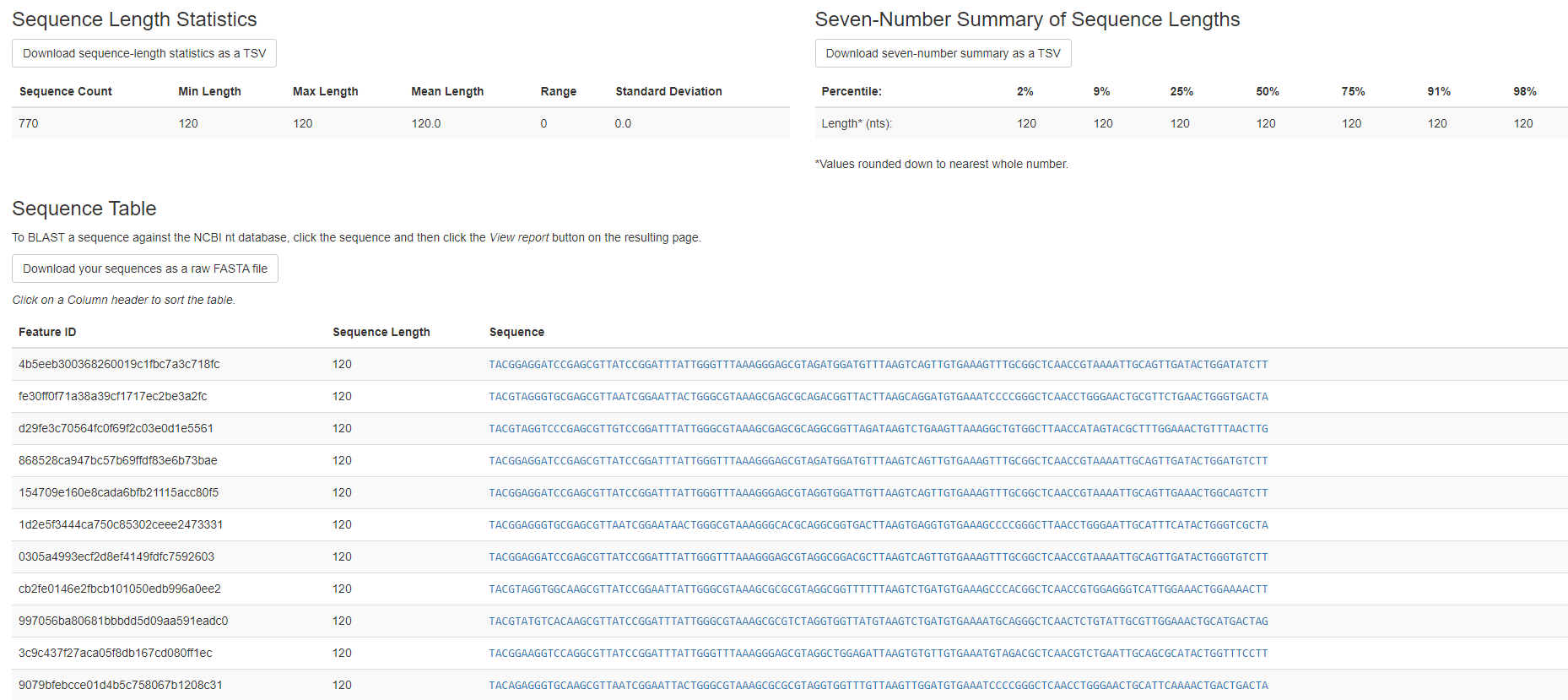
进化树构建
1
2
3
4
5
6qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences step3_dada2/rep-seqs-dada2.qza \
--o-alignment step6_tree/aligned-rep-seqs.qza \
--o-masked-alignment step6_tree/masked-aligned-rep-seqs.qza \
--o-tree step6_tree/unrooted-tree.qza \
--o-rooted-tree step6_tree/rooted-tree.qza多样性
1
qiime diversity core-metrics-phylogenetic --i-phylogeny step6_tree/rooted-tree.qza --i-table step3_dada2/table-dada2.qza --p-sampling-depth 1103 --m-metadata-file sample-metadata.tsv --output-dir step7_diversity参数1103选择的原因。
1
Here we set the --p-sampling-depth parameter to 1103. This value was chosen based on the number of sequences in the L3S313 sample because it’s close to the number of sequences in the next few samples that have higher sequence counts, and because it is considerably higher (relatively) than the number of sequences in the samples that have fewer sequences.
分组计算多样性
1
2
3
4
5
6
7
8
9qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv1
2
3
4
5
6
7
8
9
10
11
12
13qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column body-site \
--o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \
--p-pairwise
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column subject \
--o-visualization core-metrics-results/unweighted-unifrac-subject-group-significance.qzv \
--p-pairwisePCoA
1
2
3
4
5
6
7
8
9
10
11qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes days-since-experiment-start \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-days-since-experiment-start.qzv
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes days-since-experiment-start \
--o-visualization core-metrics-results/bray-curtis-emperor-days-since-experiment-start.qzv$\alpha$稀释曲线
1
2
3
4
5
6qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzv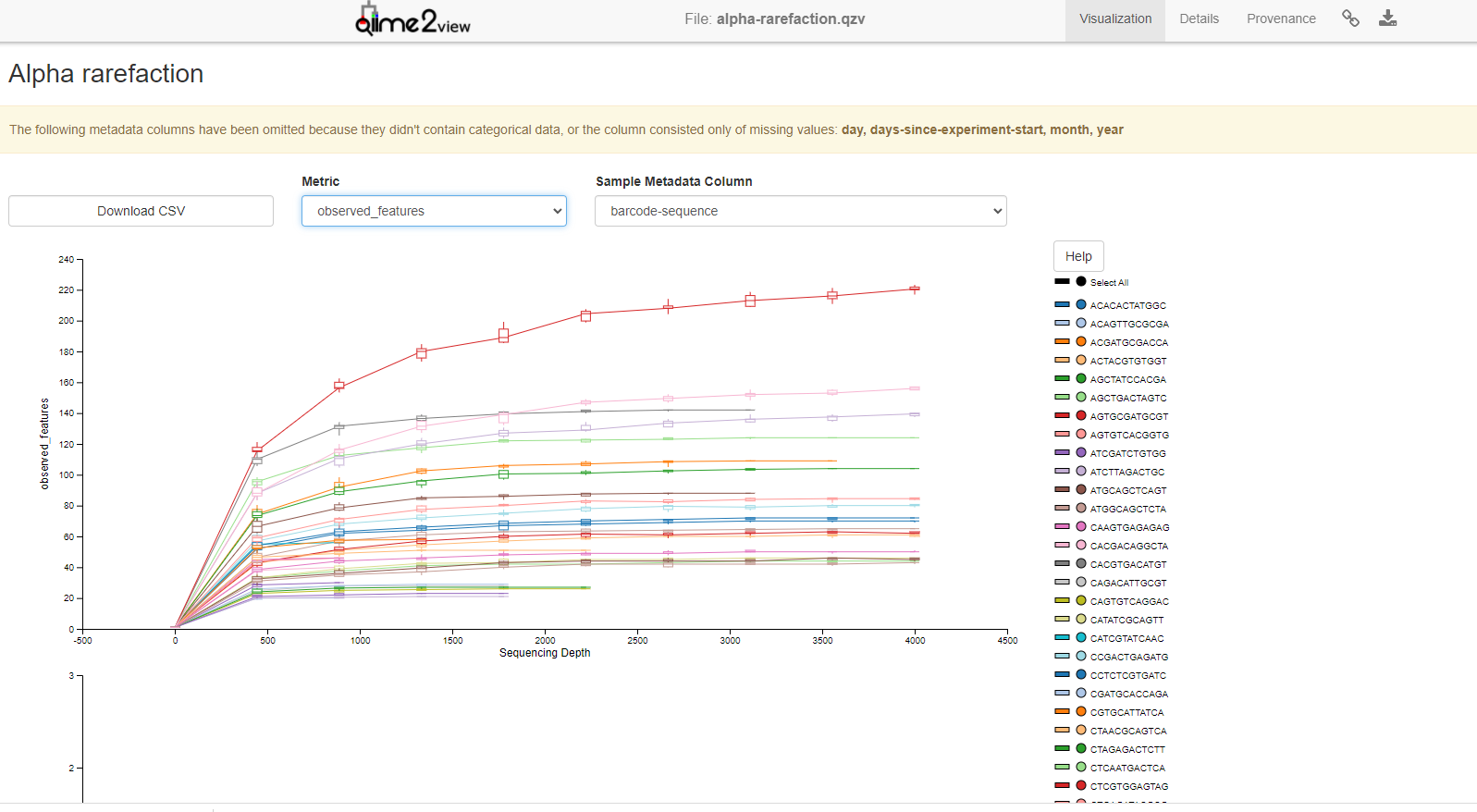
分类
下载分类器
分类
1
2
3qiime feature-classifier classify-sklearn --i-classifier classifier_db/gg-13-8-99-515-806-nb-classifier.qza --i-reads step3_dada2/rep-seqs-dada2.qza --o-classification step8_classify/taxonomy.qza
qiime metadata tabulate --m-input-file step8_classify/taxonomy.qza --o-visualization step8_classify/taxonomy.qzv
💌lixiang117423@foxmail.com
💌lixiang117423@gmail.com