文献笔记 泛基因组包含了所有的序列、基因组间的同源性和所有的变异类型。泛基因组可以用于变异检测、保守性估计、重组事件评估和推断系统发育关系。现有的方法使用reference的策略和tree-guide的方法构建泛基因组,这些方法得到的遗传变异信息会不完整,也不稳定。现有的方法基本上就是把与参考基因组足够相似的序列添加到参考基因组上,这些方法后对结构上高度可变的区域进行修剪处理,例如着丝粒和其他是卫星序列。PGGB是reference-free的流程,能够构建无偏差的泛基因组变异图。
WFMASH两两比对 PGGB从序列比对开始。reads、genes和genome甚至是这三者的组合都可以用于构建泛基因组,格式是FASTA或者是FASTQ都可以。为了避免参考基因组和顺序的偏差影响,PGGB使用all-to-all的策略进行序列比对。这样的好处就是每条序列在泛基因组中都可以作为reference来描述遗传变异。有个问题就是两两比较的话会有个问题就是需要更多的计算资源和时间,PGGB使用的是WFMASH,这个软件利用序列片段进行同源性比较,而不是使用单个碱基来进行同源性比较。这个方法能够保留较长范围内的共线性,但是对转座子和微卫星序列之间的重复序列以和较短的相似序列并不敏感。WFMASH的比对结果是PGGB构建泛基因组的理想结构,但是PGGB也可以使用任何以PAF格式存储的比对结果构建泛基因组。
SEQWISH构建variation graph 第二步是泛基因组graph构建:使用SEQWISH将基因组和成对比对结果转换为变异图(variation graph)。SEQWISH能够将上一步比对结果中的不存在的同源关系进行恢复(The SEQWISH graph recovers transitive homology relationships that may not be present in the initial alignment set)。这样的好处是能够使用随机稀疏化来降低比对结果的复杂性。首先是提取WFMASH结果中的同源比对结果,然后只align上一步mapping结果中的一个子集。我们期望泛基因组的每个同源locus的alignment graph是局部全连接的,这样的话就需要足够多的mapping. PGGB使用基于 Erdös–Rényi 随机图模型的方法来确保稀疏化的安全阈值。这样做的好处是不用计算所有的两两比对就能重建variation graph中的传递关系。这样就能在保证结果准确的前提下还能降低运行时间,增加10倍基因组,只需要增加44倍左右的时间,而不是100倍。
SMOOTHXG构建泛基因组 第三步是使用SMOOTHXG 局部压缩和简化泛基因组variation graph来完成泛基因组构建。上一步的SEQWISH构建的graph已经是近乎完整的模型,这个模型已经能够代表输入的基因组和序列之间的alignment了;但是通常会含有复杂的局部motif,这些motif会使得下游的分析变得更加复杂。之所以这样是因为输入的用于alignment的序列之间并没有进行相互标准化,从而导致低复杂度序列中变异的表示有所不同,还可能多出一些密集的小的looping motif. 为了解决这个问题,PGGB采用的方法是移除SEQWISH输入数据中短的match. 这样是可以降低复杂度,但是呢也可能会生成一个并不能代表所有局部成对关系的graph. 为了解决这个问题,PGGB使用偏序比对(partial order alignment,POA)来解决这个问题。这种方法会删除长度短于POA长度的小圈(small cycles),但是会保留更大的cycles,这些更大的cycles可以紧凑地表示结构可变的loci. 这样做有利于针对短reads、古老的和低覆盖度的基因组进行genotyping. SMOOTHXG能够使用POA的优势去优化泛基因组。GFAFFIX用于压缩非冗余的nodes,ODGI对最终的graph进行排序。
输出结果 PGGB输出的结果可以用于解释、质控和下游分析。使用ODGI可以对泛基因组进行基本的统计和可视化,也可以输出MultiQC报告。使用VG可以从PGGB输出的结果中鉴定遗传变异,生成对对应的VCF文件。为了便于后续分析,PGGB使用BiWFA将复杂的嵌套变异分解为最小化的reference-relative的表示。这时候PGGB就成为了多样本全基因组组装的遗传变异调用器。
benchmark PGGB表现和MUMMER4大差不差,但是呢MUMMER4输出的结果是针对某个参考基因组的,而PGGB输出的结果是针对所有的基因组的。
在variation graph中,nodes表示的是alleles,可以将基因组理解为等位基因计数的载体。基于这种方法可以考虑所有的变异类型,而且不会因为参考基因组的不同而有偏差。PGGB构建的系统发育树和先前的研究使用SNP构建的系统发育树一致。有趣的是,基于MUMMER结果的SNV-based构建失败的系统发育树施用PGGB构建成功。Minigraph-Cactus这种方法构建的泛基因组构建的系统发育树和先前的研究所报道的有所差异。
PGGB优势
无偏差:和序列顺序无关、和系统发育无关;
无损:输入的基因组都保留在了泛基因组中,每个基因组都可以作为下游分析的参考基因组;
模块化:每个模块输出的文件都是文本格式的,方便后续的其他泛基因组构建方法进行调用;
已验证:HPRC就是使用PGGB构建的人类泛基因组
开放性:社区开发,流程部署
安装 手动安装 需要用到的软件有:wfmash、seqwish、smoothxg、odgi和gfaffix. 这些软件可以使用conda安装。
https://github.com/waveygang/wfmash
https://github.com/ekg/seqwish
https://github.com/pangenome/smoothxg
https://github.com/pangenome/odgi
https://github.com/marschall-lab/GFAffix
选择性安装的软件有:bcftools、vcfbub、vcfwave和vg.,这几个软件用于鉴定和标准化变异;MultiQC生成简单的描述统计结果;pigz用于压缩流程中的输出文件。
https://github.com/samtools/bcftools
https://github.com/pangenome/vcfbub
https://manpages.ubuntu.com/manpages/noble/man1/vcfwave.1.html
https://github.com/vgteam/vg
https://github.com/MultiQC/MultiQC
https://github.com/madler/pigz
Docker安装 1 docker pull ghcr.io/pangenome/pggb:latest
或者从dockerhub安装:
1 docker pull pangenome/pggb
示例用法:
1 docker run -it -v ${PWD} /data/:/data ghcr.io/pangenome/pggb:latest /bin/bash -c "pggb -i /data/HLA/DRB1-3123.fa.gz -p 70 -s 3000 -n 10 -t 16 -V 'gi|568815561' -o /data/out"
-v这个参数最好是用全部的路径。
Singularity 集群上通常使用Singularity替代docker.
1 singularity pull docker://ghcr.io/pangenome/pggb:latest
下载示例数据:
1 2 git clone --recursive https://github.com/pangenome/pggb.gitcd pggb
Bioconda 1 conda install -c bioconda pggb
GUIX 1 2 3 git clone https://github.com/ekg/guix-genomicscd guix-genomics
Nextflow https://github.com/nf-core/pangenome
快速上手 泛基因组是一个物种或clade的所有序列的完整集合,能够使用variation graph这种格式来表示。为了方便交流/交换泛基因组,使用的标准格式是Graphical Fragment Assembly(GFA),现在的版本是version 1.
第一步:数据准备 将所有的fasta序列放到一个文件中,如果有很多样品和/或很多单倍型时,按照PanSN-spec规则对序列进行命名。可以选择是否使用bgzip进行压缩;然后使用SAMtools对输入文件构建索引。
1 2 3 samtools faidx in.fa
第二步:序列分割 如果有输入的数据是全基因组的组装结果,那么可以考虑按照染色体将不同的染色体进行拆分,然后使用pggb对每条染色体进行泛基因组构建。
1 2 3 4 5 6 7 partition-before-pggb -i in.fa \ input file in FASTA formatfor segmentsfor scaffolding the graph'ref:1000' make a VCF against "ref" decomposing variants >1000bp
上面的代码会生成两段独立的代码(如果有n个community的话就会生成n个下面的代码):
1 2 3 4 5 6 7 8 9 10 11 12 pggb -i output/in.fa.dd9e519.community.0.fa \
第三步:构建graph 1 2 3 4 5 6 7 8 pggb \for a seed mapping'ref:1000' make a VCF against "ref" decomposing variants >1000bp
输出的结果是outdir/input.fa*smooth.fix.gfa. 输出的结果可以使用odgi进行可视化,可以选择1D和2D这两种方式进行展示。
1D可视化
从左到右
彩色条表示二进制矩阵中嵌入路径与该泛基因组序列的分箱线性化渲染
path的名称在最左侧
路径下的黑线,即所谓的链接,表示图的拓扑结构。
2D可视化
每个彩色矩形代表路径的一个节点。节点的 x 坐标位于 x 轴上,y 坐标位于 y 轴上。
气泡表示此处某些路径具有发散序列,或者可以表示重复区域。
详细流程 序列准备 数据量较大时,如果直接把所有数据放到一起,那么就需要很大的计算量和计算资源。建议的做法是把输入数据拆分为community,简单理解就是把相同的染色体拆分到一个文件中,以减少对计算资源的消耗,降低计算压力。但是如果有染色体易位等情况时,不建议把数据进行拆分。
具体步骤 安装依赖 1 2 pip3 install python-igraph
序列命名 1 2 3 4 5 6 7 ls *.fa | while read f; do echo $f | cut -f 1 -d '.' );echo ${sample_name} "${sample_name} #1#" $f >> scerevisiae7.fastadone
重命名使用的是fastix:
https://github.com/ekg/fastix
上面这段代码所完成的工作:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 ls *.fawhile read f; do ls 的输出通过管道传递给 while 循环,每次循环读取一个文件名到变量 f 中进行处理。echo $f | cut -f 1 -d '.' );cut 命令对文件名 f 进行截断操作,以 . 为分隔符,提取其第一个字段,作为 sample_name。这一般被用于提取不带扩展名的文件名部分。echo ${sample_name} "${sample_name} #1#" $f >> scerevisiae7.fasta"${sample_name} #1#" 可能是用来指定前缀或标记以包含在输出序列中。处理结果被附加(>>)到 scerevisiae7.fasta 这个文件中。
这里将所有单倍型的编号都设置为1,是因为所用的样品是单倍体。
计算分歧度 这一步需要获取输入的组装结果之间的关系,目的是为了检测底层的community,其实也就是计算不同序列之间的相似性。
1 wfmash scerevisiae7.fasta.gz -p 90 -n 6 -t 4 -m > scerevisiae7.mapping.paf
-p设位置为90是因为期望的序列之间的分歧度是10%左右,分歧度的估计参考下一节内容,-n表示单倍型的数量,最小值是1.
paf格式转network格式 接下来是将paf格式的结果转换为网络格式,使用脚本paf2net.py:
1 python3 ../../scripts/paf2net.py -p scerevisiae7.mapping.paf
这个脚本生成三个文件:
1 2 3 4 5 scerevisiae7.mapping.paf.edges.list.txt is the edge list representing the pairs of sequences mapped in the PAF;'id to sequence name' map.
紧接着运行下面的代码检测community:
1 2 3 4 python3 ../../scripts/net2communities.py \
paf2net.py这个脚本会生成一系列的.community. .txt,每个文件里面的序列就是一个community,每一行是一条序列。
1 cat scerevisiae7.mapping.paf.edges.weights.txt.community.6.txt
示例输出:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 DBVPG6044
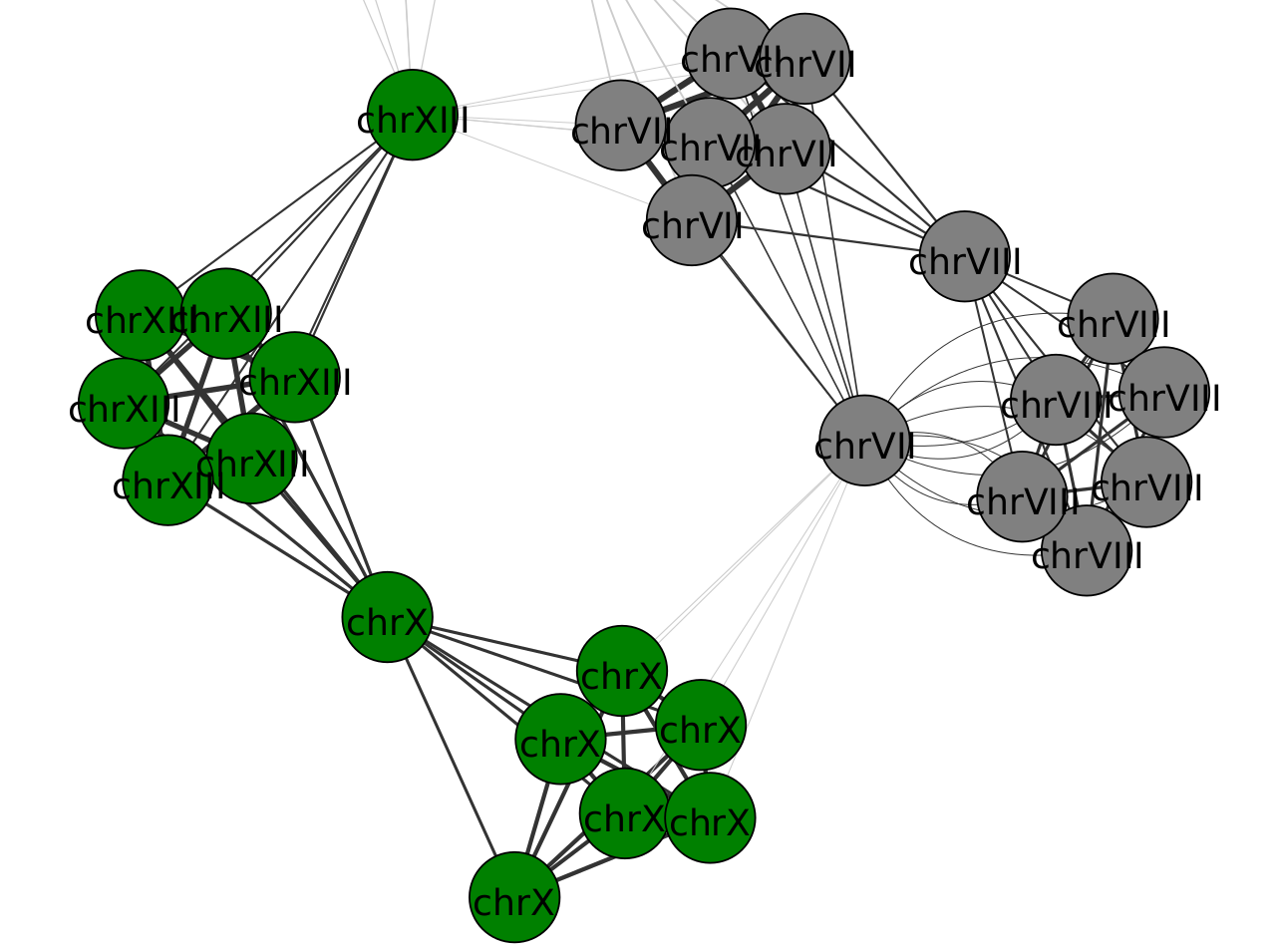
这个结果之所以会 chrVII和chrVIII是因为这些样品中有染色体重排现象。
想查看每个community中的序列信息,执行下面的代码:
1 2 3 4 seq 0 14 | while read i; do cat scerevisiae7.mapping.paf.edges.weights.txt.community.$i .txt | cut -f 3 -d '#' | sort | uniq | tr '\n' ' ' );echo "community $i --> $chromosomes " ;done
示例输出:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 community 0 --> chrI
可以看到community 6和community 8这两个community有多个染色体。
1 2 3 4 5 python3 ../../scripts/net2communities.py \
数据拆分 鉴定完community后可以使用pggb进行管理。执行下面的代码拆分每个community:
1 2 3 4 5 6 seq 0 14 | while read i; do echo "community $i " cat scerevisiae7.mapping.paf.edges.weights.txt.community.$i .txt) | \$i .fa.gz$i .fa.gzdone
生成的scerevisiae7.community.*.fa.gz文件现在就可以作为pggb的输入文件。如果需要整合所有的community的graphs,可以使用odgi squeeze.
基于Mash的拆分方法 直接使用mash计算序列之间的距离:
1 mash dist scerevisiae7.fasta.gz scerevisiae7.fasta.gz -s 10000 -i > scerevisiae7.distances.tsv
-s设置得越大,得到的结果也越准确;-i参数表示比较输入文件内的序列,而不是比较整个输入文件。
查看结果:
1 2 3 4 5 6 head -n 5 scerevisiae7.distances.tsv | column -t
第三列表示mash距离,第四列是P值,第五列共享哈希值的数量除以所考虑哈希值的数量的比率。
也可以将mash距离转换为network格式,进一步坚定community:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 python3 ../../scripts/mash2net.py -m scerevisiae7.distances.tsvseq 0 14 | while read i; do cat scerevisiae7.distances.tsv.edges.weights.txt.community.$i .txt | cut -f 3 -d '#' | sort | uniq | tr '\n' ' ' );echo "community $i --> $chromosomes " ;done
输出的结果和上一个方法是一致的。
分歧度估计 pggb中的一些参数会影响最终构建的graph的结构。例如,-p参数影响的是mapping identity,这个参数设置得越小,对比的灵敏度就会随之增加,就会得到更多压缩后的graphs. 这个参数的设置依赖于输入序列的分歧程度。
详细步骤 下载基因组 1 2 3 mkdir -p assemblies/yprp_panelcd assemblies/yprp_panelcat ../../docs/data/scerevisiae.yprp.urls | parallel -j 4 'wget -q {} && echo got {}'
将基因组序列和线粒体序列合并:
1 2 3 4 ls *.fa.gz | cut -f 1 -d '.' | uniq | while read f; do echo $f $f .* > $f .fadone
重命名序列 1 2 3 4 5 6 7 ls *.fa | while read f; do echo $f | cut -f 1 -d '.' );echo ${sample_name} "${sample_name} #1#" $f >> scerevisiae7.fastadone
序列分歧度 假设一次只对一条染色体进行泛基因组构建,就需要估计染色体之间的分歧程度。使用下面的代码:
1 2 3 4 5 6 cut -f 1 scerevisiae7.fasta.gz.fai | cut -f 3 -d '#' | sort | uniq | while read CHROM; do $CHROM .fasta.gz"$CHROM \t" scerevisiae7.fasta.gz.fai | cut -f 1) | bgzip -@ 4 > $CHR_FASTA echo "Generated $CHR_FASTA " done
计算分歧度的方法就是依次进行比较。mash triangle数据一个三角矩阵的结果,类似于相关性那种结果。
1 2 mash triangle scerevisiae7.chrMT.fasta.gz > scerevisiae7.chrMT.mash_triangle.txtcat scerevisiae7.chrMT.mash_triangle.txt | column -t
输出的结果类似于:
1 2 3 4 5 6 7 8 7
上面的结果可以看出这个community中有7条序列,mash距离从0到1之间。使用下面的代码计算最大分歧度:
1 sed 1,1d scerevisiae7.chrMT.mash_triangle.txt | tr '\t' '\n' | grep chr -v | LC_ALL=C sort -g -k 1nr | uniq | head -n 1
输出结果为0.0210181. 这个结果是一个community的mash分歧度,使用下面的代码计算所有community的最大分歧度:
1 2 3 4 5 6 7 8 ls scerevisiae7.*.fasta.gz | while read CHR_FASTA; do echo $CHR_FASTA | cut -f 2 -d '.' )$CHR_FASTA | sed 1,1d | tr '\t' '\n' | grep chr -v | LC_ALL=C sort -g -k 1nr | uniq | head -n 1)echo -e "$CHROM \t$MAX_DIVERGENCE " >> scerevisiae7.divergence.txtdone cat scerevisiae7.divergence.txt | column -t
输出结果为:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 chrI 0.0178312
从结果中可以看出chrVII、chrVIII和chrXIII这三条染色体的分歧度排在前面,最大值是0.0639874. 那到底如何设置mapping identity也就是-p这个参数呢?通常的原则是100 - max_divergence * 100,这个数据中应该设置为93.60126,建议是等于或小于这个值。但是,为了解释那些被低估的序列分歧度,或者为了避免局部序列中较大或适中的结构变异造成的序列分歧差异,建议把这个参数设置为更小的值,比如90.
染色体间的分歧度估计 如果样品之间有染色体重排或者是易位等现象,那就需要计算染色体之间的序列分歧度。
1 2 mash triangle scerevisiae7.community.0.fa.gz -s 10000 > scerevisiae7.community.0.mash_triangle.txtcat scerevisiae7.community.0.mash_triangle.txt | column -t
输出结果为:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 14
输出结果中chrVII和chrVIII这两条染色体,因为前期的研究发现只有这两条染色体有重排现象。-s设置为10000是为了提高比对的准确度。较低的分歧度很大程度上是因为发生了染色体结构重排。
小变异鉴定 主要组织相容性复合体 (MHC) 是脊椎动物 DNA 中的一个大型位点,包含一组紧密相连的多态性基因,这些基因编码对适应性免疫系统至关重要的细胞表面蛋白。在人类中,MHC 区域位于 6 号染色体上。
详细步骤 数据收集 将chromosome 6 contigs比对到MHC ALT的单倍型上。
1 wfmash -t 16 -m -s 5000 -p 98 MHC.fa.gz chr6.pan.fa > HPRCy1.pan.chr6_vs_MHC.paf
然后根据比对结果构建一个BED文件:
1 2 awk '{ print $1, $3, $4 }' HPRCy1.pan.chr6_vs_MHC.paf | tr ' ' '\t' | sort -V > HPRCy1.MHC.bed
最后一步是提取对应区域的序列:
1 2 bedtools getfasta -fi /lizardfs/erikg/HPRC/year1/parts/chr6.pan+refs.fa -bed HPRCy1.MHC.merge.bed | bgzip -c -@ 16 > HPRCy1.MHC.fa.gz
序列重命名 前面的处理已经保证名称的统一,直接使用即可:
1 2 3 4 5 6 7 cut -f 1 HPRCy1.MHC.fa.gz.fai | head -n 6
分歧度估计 由于这个数据量很小,就省去了计算分歧度这一步。
构建泛基因组 1 pggb -i HPRCy1.MHC.fa.gz -p 95 -n 90 -t 16 -G 13117,13219 -o HPRCy1.MHC.s10k.p95.output
graph统计 1 2 3 odgi stats -i HPRCy1.MHC.s10k.p95.output/*.final.gfa -t 16 -S
使用vg鉴定突变 1 2 vg deconstruct -P chm13 -H '?' -e -a -t 48 HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.gfa | \
-H参数设置为’?’是为了path的名称,提取每个contigs的变异情况。
下一步是过滤变异并重新排列参考/突变的alleles:
1 2 3 vcfbub -l 0 -a 100000 --input HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.chm13.vcf.gz | \
从PGGB的VCF文件提取SNP:
1 2 3 4 5 6 7 8 9 10 11 12 REF=chm13cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do echo $CONTIG $CONTIG \$REF done
使用nucmer鉴定突变 首先准备序列:
1 samtools faidx HPRCy1.MHC.fa.gz chm13
然后将序列必读到参考基因组:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 REF=chm13mkdir -p nucmer/cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do echo $CONTIG ${CONTIG} _vs_${NAMEREF} $CONTIG > $CONTIG .fa$REF $CONTIG .fa --prefix "$PREFIX " "$PREFIX " .delta > "$PREFIX " .var.txtrm $CONTIG .fadone
再将nucmer的结果转换为vcf格式:
1 2 3 4 5 6 7 8 9 10 11 12 REF=chm13"4.0.0beta2" cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do echo $CONTIG ${CONTIG} _vs_${NAMEREF} "$PREFIX " .var.txt $CONTIG $REF $NUCMER_VERSION $PREFIX .vcf$PREFIX .vcf$PREFIX .vcf.gzdone
变异评估 将参考序列转换为SDF格式:
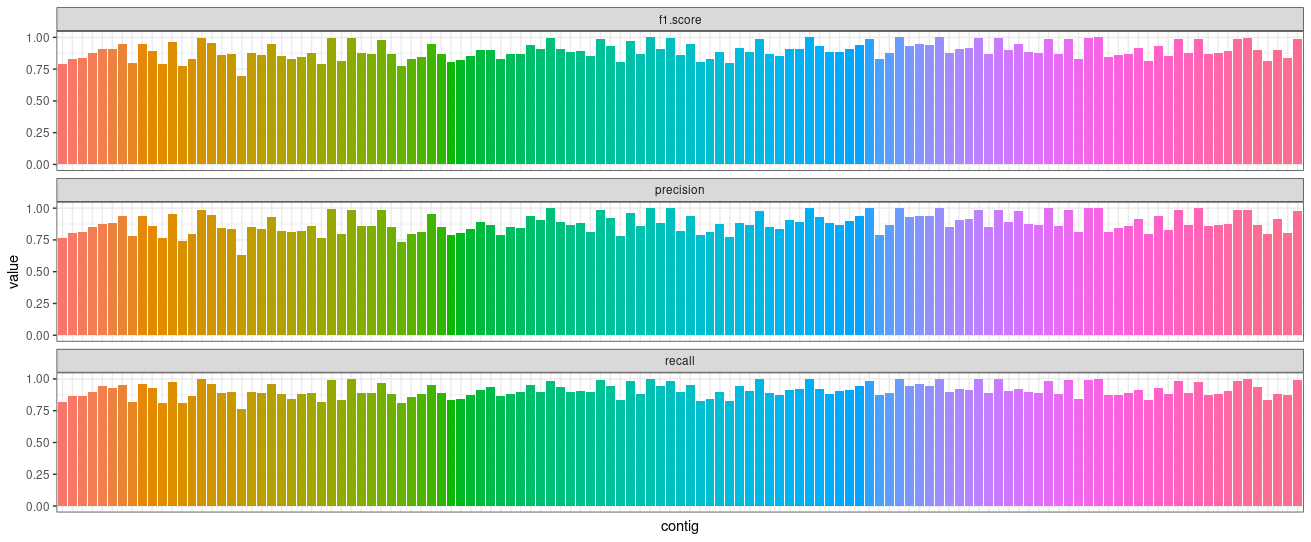
然后比较PGGB和nucmer两个方法鉴定到的SNP:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 REFSDF=chm13cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do echo $CONTIG ${CONTIG} _vs_${NAMEREF} ${CONTIG} .max1.vcf.gzwhere to evaluate the variants$REFSDF \$PREFIX .vcf.gz \$PATH_PGGB_VCF \$dist -i $PREFIX .vcf.gz ) -b <(bedtools merge -d $dist -i $PATH_PGGB_VCF )) \${CONTIG} done
获取统计结果:
1 2 cd vcfevalecho contig precision recall f1.score; grep None */*txt | sed 's,/summary.txt:,,' | tr -s ' ' | cut -f 1,7,8,9 -d ' ' ) | tr ' ' '\t' > statistics.tsv
最后使用R语言绘图:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 require(ggplot2)'statistics.tsv' , sep = '\t' , header = T, comment.char = '?' )"Metric" )stat ="identity" ) +"none" )
重要的参数 Mapping 在pggb中,对泛基因组graph结构影响最大的是mapping iedntity参数-p和片段长度-s. 这两个参数传给wfmash后对泛基因组的基础结构的影响非常大。
当泛基因组比较小,或者是重复序列比较少时,-s这个参数可以设置小一些。更大的-s参数能够保证尽可能多的保留输入序列中的共线性区域,通常需要大于转座子和其他常见的重复的长度。在wfmash中默认只保留至少mapping了5次的segment,可以使用参数-l, —block-length BLOCK进行调整。
-p默认的参数是95,-s默认的参数是10000. 但是还是要根据实际情况来调调整这两个参数。如果是分歧度很高的样品群,那么就需要把-p和-s调整到不同的水平。增加这两个参数的话,会增加初始比对的时的严格程度;降低这两个参数呢会使得比对时的敏感性更高。
还有一个参数是-n,表示的是单倍型的数量,这个在执行PanSN-spec流程的时候会自动计算。
分歧度估计 使用mash dist或者是mash triangle来计算序列之间的分歧度,通过输出的结果调整-p参数,通常是100-100*最大的分歧度数值。
Target number of alignment pggb构建的泛基因组由每个基因组每个片段的mapping数决定,对应的参数是-c. 理想情况是设置为1,也就是每条序列都两两比较。但是,如果泛基因组中有拷贝数变异,那么-c这个参数就应该设置得更大一些。一个原则是:比对的次数与N*N成正比,而且有些多重mapping可能是冗余的。如果没有特殊的考虑,直接设置为1即可。
match filter设置 当我们从输入的比对结果中把很短的match过滤掉时,得到的graph结合seqwish会有更好的表现结果。wfmash比对结果中的这些区域通常是具有较低比对质量的区域,通常是大片段的indels和结构变异。最后的smoothxg step这一步中会得到解决。这个过滤的方法对应的参数是-k. 默认的值是23,这时候的偏差可以达到5%左右。当序列分歧度较大时,增大-k的值;当序列的分歧度较小时,降低-k的值。人类的单倍型基因组可以把-k设置为79. 那么这个参数是什么意思呢?-k设置为N就表示可以容忍不超过1/N的局部差异比例。可以使用minimap2等方法来减少这个步骤的影响,这种方法是通过更好地展示输入的比对文件中的大片段indels实现的。
Homogenizing and ordering the graph pggb的最后一步是细化graph,这种细化是通过POA来实现的。这个步骤会涉及到3个参数:-G、-P和-O. -G参数在理想情况下可以设置为泛基因组中转座子重复序列的长度,可以大于这个值,这个值设置得越高,得到的泛基因组的质量就会更好一些;更高的-G参数会得到多样性更低的泛基因组。minimap2对-P这个参数给出了一些建议:
asm mode **--poa-params**divergence in %
asm5
1,19,39,3,81,1
~0.1
asm10
1,9,16,2,41,1
~1
asm20
1,4,6,2,26,1
~5
Variant Calling -V参数指定Reference.
Reporting -S生成graph统计,-m生成MultiQC报告,-v这个参数如果没有设置的话,会同时生成1D和2D的绘图结果。
Parallelization -t参数指定运行的线程数,通常是一半即可。smoothxg步骤中的POA可以设置对应的-T.
更大的泛基因组 还是关注-p和-s这两个参数。-s这个参数通常设置为5-20 kbp,-p设置为接近序列分歧度的那个值计算得到的值。如果想获得高质量的比对结果,就把-p设置大一些。增加-s和-p会减少运行的时间以及内存需求,但是会降低graph的压缩程度。
如果使用10-20个基因组,多样性在1-5%,那么这两个参数就可以设置为-s10000 -p 95;但是,如果想把其他物种的基因组也加进这个泛基因组(多样性20%),那么-p这个参数就应该设置为70.
使用SPOA或abPOA处理复杂的blocks时,需要大量的内存。可以设置-T参数来降低内存的消耗。
示例文献 葡萄泛基因组 https://www.nature.com/articles/s41588-024-01967-5
To represent the genetic diversity of grapes, we constructed a graph pangenome that incorporated a total of 29 haplotype assemblies, including 18 newly sequenced haplotype assemblies from 9 phase-resolved accessions and 11 previously published assemblies based on continuous long read data (two phase-resolved and seven primary assemblies). Based on these genome assemblies, we used two tools to build graph pangenome, MC (v.2.6.11) and PGGB (v.0.5.4), respectively. We combined reads mapping and assembled alignments to validate the graph paths, edges and nodes in the grape pangenome. To address the issue of small fragments in assemblies, we implemented filtering steps to exclude minor, fragmented and diverse assemblies that could introduce the wrong structures.
野生和驯化大麦 https://www.nature.com/articles/s41586-024-08187-1
The coordinates of amy1_1 copies in 76 genome assemblies were obtained by BLAST searches with the Morex allele of HORVU.MOREX.PROJ.6HG00545380. The genomic intervals surrounding amy1_1 from 10 kb upstream of the first copy to 10 kb downstream of the last copy were extracted from corresponding assemblies and used for further analyses. We applied PGGB (v.0.4.0, https://github.com/pangenome/pggb ) for 76 amy1_1 sequences with parameters ‘-n 76 -t 20 -p 90 -s 1000 -N’. The graph was visualized using Bandage125 (v.0.8.1). ODGI (v.0.7.3, command ‘paths’)110 was used to get a sparse distance matrix for paths with the parameter ‘-d’. The resultant distance matrix was plotted with the R package pheatmap (https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf ). Six representative sequences of amy1_1 were aligned against Morex by BLAST+ (v.2.13.0).
马铃薯泛基因组 https://www.nature.com/articles/s41586-024-08476-9
The Minigraph-Cactus pipeline27 and PGGB26 were used to construct a pseudo-phased pangenome with all 61 haplotypes based on the whole-genome alignment (including the DMv6.1 reference genome). For the PGGB, we estimated the divergence of each chromosome with mash distances and confirmed chromosome community with wfmash87 mapping. Then, we used “pggb -s 10000 -n 61 -p 90 -k 47 -P asm20 -O 0.001” to build each chromosome graph. We visualized the 1D layout of the graph and estimated presence and absence ratios to the DMv6.1 reference in 100-kb sliding windows using ODGI88. The small variants and SVs were detected by vg deconstruct from snarls, and we only kept top-level and <1-Mb variants with vcfbub. For the Minigraph-Cactus pipeline, we assigned DMv6.1 as the guide for the paths, and progressively aligned the 60 haplotypes to it. We used the cactus-pangenome script with parameters “—gfa full —gbz full —vcfReference DMv6.1” to generate complete workflows and execute commands. The generated graph fragment assembly (GFA) format graph was used for edge, node and coverage statistics and subgraph generation from a BED input. The VCF output file comprises all graph variations based on the DMv6.1 reference, enabling the calculation of polymorphisms.
结构变异挖掘综述 https://www.cell.com/cell/abstract/S0092-8674(24)00004-7?rss=yes
人类泛基因组 https://www.nature.com/articles/s41586-023-05896-x