下载安装
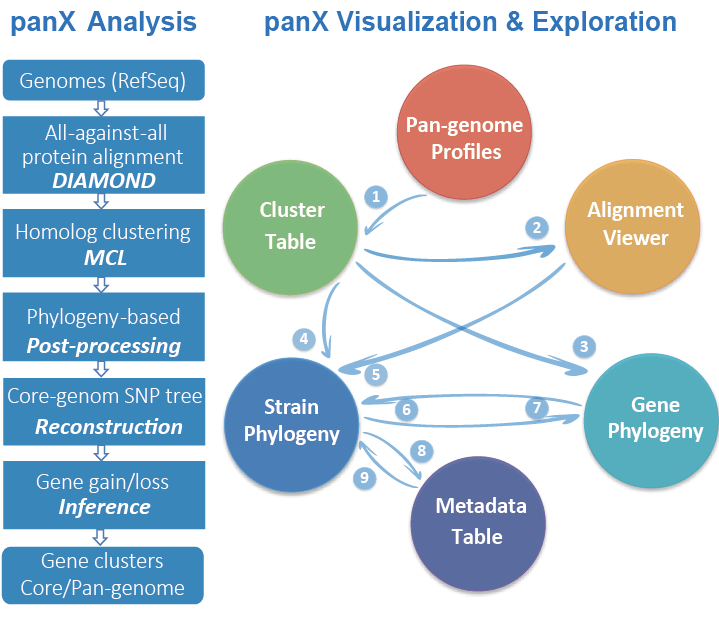
Ding W, Baumdicker F, Neher R A. panX: pan-genome analysis and exploration[J]. Nucleic acids research, 2018, 46(1): e5-e5.
我是用mamba安装的:
1 2 3 mamba create --name panX
然后依次安装这些软件就好:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 - python==2.7.*
然后从官方地址 把项目克隆或者下载下来就好。
数据准备 官方的示例数据是NCBI的gbk格式,可是通常都是没有这种格式的,就在我要放弃的时候发现一个参数:`-ngbk,当没有gbk文件的时候加上这个参数即可。
需要的数据有基因核酸序列和蛋白序列,而且两个文件的文件名称以及序列的ID必须完全一致,不然会报错! 核酸序列后缀是fna,蛋白序列的后缀是faa.
由于是使用DIAMOND进行序列比对,因此在蛋白序列中不能出现.,ChatGPT帮忙写了个脚本去除蛋白序列中的.:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 from Bio import SeqIOimport sys1 ] 2 ] with open (output_file, "w" ) as output_handle:"fasta" )for seq in input_sequences:if "." in seq.seq:if seq.seq.index("." ) == len (seq.seq)-1 :1 ]"fasta" )elif "." in seq.seq[1 :-1 ]:continue else :"fasta" )else :"fasta" )
开始运行 将核酸序列和蛋白序列放在一个文件夹下,加入我的文件名叫project,那就将核酸序列和蛋白序列全部放在这个文件夹下,然后进入这个文件夹,然后运行:
1 /path/to/panX/panX.py -fn ./ -sl bin.2 -ngbk -t 65
fn表示文件夹路径,如果和核酸蛋白文件在一个目录就用./;sl参数表示临时文件的名称;-ngbk表示输入文件不是gbk格式;-t:线程,越多越快。
运行后就会有这些文件和文件夹:
其他 部分运行日志:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Running panX in main folder: /home/u ser/project/ sanqi.metagenomics/14.bins.pangenome/ bin.2 /'/home/user/project/sanqi.metagenomics/14.bins.pangenome/bin.2/*.faa' 调用 stat 失败: 没有那个文件或目录from GenBank file for step03:0.36 minutes (21.55 seconds) from GenBank file for step04:0.00 minutes (0.00 seconds) 0.03 minutes (1.67 seconds)/home/u ser/mambaforge/ envs/panX/ bin/diamond makedb -p 65 --in / home/user/ project /sanqi.metagenomics/ 14 .bins.pangenome/bin.2/ protein_faa/diamond_matches/ reference.faa -d /home/u ser/project/ sanqi.metagenomics/14.bins.pangenome/ bin.2 /protein_faa/ diamond_matches/nr_reference> / home/user/ project /sanqi.metagenomics/ 14 .bins.pangenome/bin.2/ protein_faa/diamond_matches/ diamond_makedb_reference.log 2 >&1 20.64 minutes (1238.37 seconds)600 /home/u ser/mambaforge/ envs/panX/ bin/diamond blastp --sensitive -p 65 -e 0.001 --id 0 --query-cover 0 --subject-cover 0 -k 600 -d / home/user/ project /sanqi.metagenomics/ 14 .bins.pangenome/bin.2/ protein_faa/diamond_matches/ nr_reference -f 6 qseqid sseqid bitscore -q /home/u ser/project/ sanqi.metagenomics/14.bins.pangenome/ bin.2 /protein_faa/ diamond_matches/reference.faa -o / home/user/ project /sanqi.metagenomics/ 14 .bins.pangenome/bin.2/ protein_faa/diamond_matches/ query_matches.m8 -t ./ > / home/user/ project /sanqi.metagenomics/ 14 .bins.pangenome/bin.2/ protein_faa/diamond_matches/ diamond_blastp_reference.log 2 >&1