泛基因组工具vg
vg:variation graph data structures, interchange formats, alignment, genotyping, and variant calling methods.
![]()
1 基础知识
vg其实就是variation graph的缩写:),一个vg中有这些元素:
1.1 **Node**(节点)
定义:表示基因组中的一段序列片段,可以是:
核心基因(所有个体共享的序列)。
- 可变基因或结构变异(如插入、缺失、倒位等)。
单核苷酸多态性(SNP)的不同等位形式。
特点:
每个节点可能对应多个等位(allele),例如不同个体在同一位置的变异。
- 节点之间可以是连续的(如参考基因组的线性序列),也可以通过边连接非连续的变异区域。
示例:
在人类基因组中,一个节点可能代表某段DNA序列(如基因区域),而另一个节点可能代表该区域的SNP变异(如A→T)。
1.2 **Edge**(边)
- 定义:表示节点之间的连接关系,描述序列如何从一个节点过渡到另一个节点。
特点:
边可以是物理相邻的连接(如参考基因组中相邻的序列)。
- 也可以表示变异路径(如不同等位或结构变异之间的选择)。
- 边可能带有权重(如变异频率)或标签(如变异类型)。
示例:
如果一个节点代表插入序列,另一个节点代表缺失,边可以表示两种变异之间的连接关系(如某个群体更倾向于携带插入)。
1.3 **Path**(路径)
- 定义:表示某个个体基因组在泛基因组图中的行走路径,由一系列节点和边组成,反映该个体的独特基因组序列。
特点:
不同个体的路径可能不同,体现遗传多样性。
- 路径可以覆盖核心基因(所有个体共享)和可变基因(部分个体特有)。
示例:
个体A的路径可能是:节点1(参考序列)→ 节点2(SNP等位A)→ 节点3(插入序列),而个体B的路径可能是:节点1→节点2(SNP等位T)→节点4(无插入)。
1.4 三者关系与泛基因组的意义
- 整合多样性:通过节点和边整合所有已知变异,避免传统线性参考基因组的单一性偏差。
- 灵活表示:路径允许不同个体的基因组以不同方式遍历图结构,准确反映变异组合。
应用场景:
序列比对:将测序数据比对到泛基因组图,提高变异检测的准确性。
- 进化分析:通过路径差异推断群体结构或进化关系。
- 功能研究:关联特定路径(如携带某结构变异的路径)与表型。
1.5 类比解释
将泛基因组图想象成地铁网络:
- 节点 = 地铁站(不同的序列位置或变异)。
- 边 = 轨道(连接站点的路线,代表可能的序列过渡)。
- 路径 = 乘客的行程(个体基因组选择的具体路线,反映其遗传组成)。通过这种结构,泛基因组能够更全面、灵活地表示物种的遗传多样性,为精准基因组学分析提供基础。
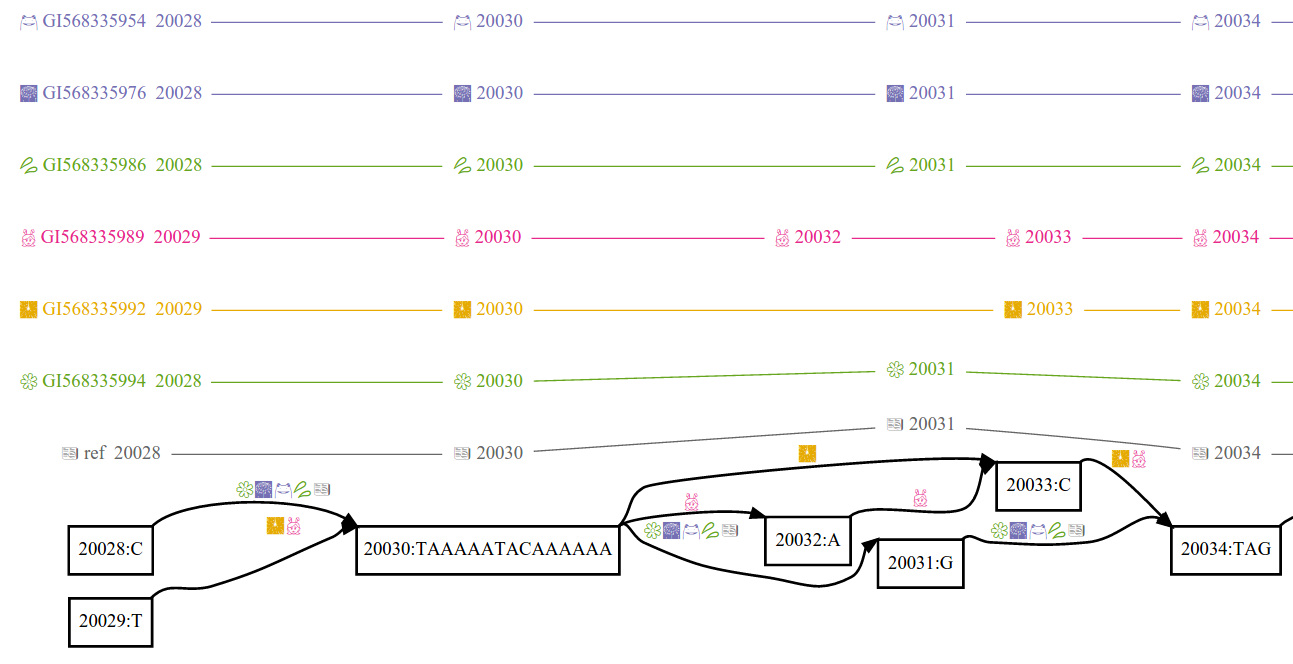
如何通俗地理解vg这种泛基因组格式呢?下图可以很容易地看出来这种格式的特点。所有的序列都可以看成node,也就是一个“点”,这个点可以是一个SNP,也可以是一大段片段。如果某个node在所有的样品里面都存在,那么这些node就会都连接到这个node,如下图中的20030.;如果是单个存在的,就用一条单独的edge连接这个node和下一个node,就像下图黄色的这个序列一样,直接从20030→2033,也就是缺失了20031和20032这两个nodes;如果多个样品共享一个node的,只用一个edge即可,就像2009→20030一样。
1.6 vg的优势
使用paths可以保证每条序列都是唯一的,当有新的序列被添加进去以后不会影响原理的序列的空间坐标,保证了结果的稳定性。
2 安装方法
Linux系统最好的安装方式就是使用编译好的版本,下载链接:
https://github.com/vgteam/vg/releases/latest
然后添加可执行权限即可:
1 | |
测试是否安装成功:
1 | |
输出下面的内容就是安装成功了:
1 | |
如果使用conda安装PGGB的话01.PGGB,会自动安装上最新版本的vg.
3 快速上手
3.1 vg构建
3.1.1 从VCF文件构建
这种方法直接使用参考基因组和VCF文件进行构建,就两个参数:
1 | |
需要注意的是需要使用SAMtools中的工具tavix对VCF文件构建索引:
1 | |
3.1.2 从组装结果构建泛基因组
可以直接使用组装后的基因组进行泛基因组组装,所用的方法是 Minigraph-Cactus:
https://github.com/ComparativeGenomicsToolkit/cactus/blob/master/doc/pangenome.md
3.2 导入和导出不同格式的graph
3.2.1 vg支持的泛基因组格式
PackedGraph(.vg):这种格式是vg的原生格式,优点是可以编辑,但是样品数量足够多或者是泛基因组太复杂时效率不足,尤其是path较多时。GFA(.gfa):GFA是一种标准的基于文本的格式,是vg和其他泛基因组工具之间处理数据时最好的格式。vg可以直接对未压缩的GFA文件进行操作,是PackedGraph将泛基因组导入到内存中进行操作的。GBZ(.gbz):GBZ是一种高度压缩的格式,可以使用较少的空间存储更多的paths,但是呢不利于编辑等操作。
可以使用下面的命令查看泛基因组的格式:
1 | |
3.2.2 导入泛基因组
通常情况下可以使用vg autoindex从GFA或者是VCF文件构建泛基因组并构建索引,或者是通过Minigraph-Cactus构建泛基因组并建立索引。此外,还可以使用vg convert -g从ODGI或PGGB等工具输出的结果中导入GFA格式的泛基因组。
3.2.3 导出泛基因组
可以使用vg convert -f将任意格式的grapha转换为GFA格式。vg默认使用V1.1的GFA,paths用W-lines表示;如果使用P-lines的会需要V1.0的GFA,代码是vg convert -fW.
3.2.4 Path类型
GBZ格式可以区分REFERENCE和HAPLOTYPE这两种paths. REFERENCEpath可以作为坐标系统,但是需要更多的资源来存储结果。HAPLOTYPE这种path类型是高度压缩的,但是不能用于位置定位查找。在人类泛基因组中(HPRC),来自GRCh38和CHM13的contigs是REFERENCEpath,其余样品的contigs是HAPLOTYPEpath.
这两种path的区别被延续到.vg和.gfa格式中,以便想换转换和操作。在.gfapath中,REFERENCEpath是P-lines或者是W-lines,对应的样品名称放在了好header中。但是在HPLOTYPEpath的W-lines中,样品名称没有在header中。 在.vg中使用特定的命名约定来表示。
需要注意的是,如果想在.vg中加载大规模的HAPLOTYPEpaths,只能用GBZ格式。vg convert -H可以把HAPLOTYPEpaths移除。
3.3 查看graph
使用vg view可以把graph转换为多种格式:
1 | |
3.4 Mapping
vg中有多个模块可以用于mapping:
vg giraffe:适合高准确度的短reads,将这些reads比对到graph上,能够保留单倍型信息。vg map:常用的mapper.vg mpmap:可以实现multi-pathmapping,以便对不确定的局部alignment进行描述。这种方法尤其适用于转录组数据。
3.4.1 vg giraffe
使用vg giraffe的第一步是构建索引,最佳方式是vg autoindex. 为了保证vg autoindex能够使用VCF文件中的单倍型信息,可以把VCF文件和对应的线性参考基因组同时给vg autoindex.
1 | |
3.4.2 vg map
如果泛基因组的graph表达,那么就需要使用vg index来存储graph,然后使用vg map进行比对。所构建的索引包括graph和特定大小的kmers. 在mapping的时候,可以使用任何比构建索引所用的kmers;当使用的kmers没有成功mapping时,会自动降低kmers来增加mapping的灵敏度。
1 | |
3.5 Graph augmentation
在mapping鉴定到的variation可以反向添加到graph中,这个过程叫做Augmentation(增强)。但是需要注意的是vg augment用于变异鉴定的话还处于试验阶段,不稳定,官方的结果是这种方法不如DeepVariant等线性变异鉴定工具。
Warning Using vg augment for variant calling remains very experimental. It is not at all recommended for structural variant calling, and even for small variants, you will often get much more accurate results (at least on human) by projecting your alignment to BAM and running a linear variant caller such as DeepVariant.
1 | |
3.6 Variant Calling
3.6.1 read support
1 | |
默认情况下vg call会忽略0/0这种类型的变异,这样可以让生成的VCF文件更加紧凑,但是不利于比较不同样本之间的差异。即使使用同一个graph得到的VCF文件也难以比较,因为坐标不同。-a参数可以解决这个问题,添加这个参数之后会对每个snarl进行比对,这时候使用的是相同的坐标体系。输出的结果可以使用bcftools merge -m all进行合并。
1 | |
如果要考虑reads中新的变异的话,就需要使用augmented后的graph哥GAM.
1 | |
可以使用类似的步骤对VCF文件中的已知的变异进行基因分型,有个要求是graph必须是使用vg construct -a构建的,vg autoindex和Minigraph-Cactus构建的graph不能用。
1 | |
在Calling SNP之间可以先对GAM进行预过滤处理:
1 | |
如果graph较大,那么建议将snarls分开进行计算:
1 | |
注意:vg augment、vg pack、vg call和vg snarls可以对任意格式的graph进行处理,包括.gfa、.vg、.xg;还能处理vg convert转换后的graph;但是不能处理augment处理后的graph. .vg和.gfa组要最多的内存,graph较大时不建议使用这两种格式。vg pack输出的结果只能和用于构建它的grap一起施用,所以vg pack x.vg -g aln.gam -o x.pack和vg call x.xg -k x.pack不能正常运行。
3.6.2 Calling variants from paths in the graph
一个从利用多个基因组比对结构构建的graph中Calling SVs的示例:
1 | |
基于.gbz或.gbwt索引的Haplotype paths可以分别使用参数-z和-g指定。使用vg call遇到较大的graph推荐将每个snarls分开计算,使用-r参数进行传参。
3.7 转录组分析
vg有一些工具可以用于转录组分析,需要用到的是剪切过的graph,也就是将带有注释的剪切点以edges的格式添加到graph中,用到的命令是vg rna. 然后可以使用vg mpmap进行比对。针对单倍型比对定量的工具是rpvg. 最简单是反式就是使用vg autoindex为vg mpmap构建索引。
- 首先构建剪切graph:
1 | |
- 然后进行比对:
1 | |
3.8 Alignment
如果graph较小,那么可以使用偏序比对将一条序列比对到graph上:
1 | |
Note that you don’t have to store the graph on disk at all, you can simply pipe it into the local aligner:
1 | |
4 常用命令
autoindex: construct graphs and indexes for other tools from common interchange file formatsconstruct: graph constructionindex: index features of a graph in a disk-backed key/value storemap: map reads to a graphgiraffe: fast, haplotype-based mapping of reads to a graphmpmap: short read mapping and multipath alignment (optionally spliced)surject: project graph alignments onto a linear referenceaugment: add variation from aligned reads into a graphcall: call variants from an augmented graphrna: construct splicing graphs and pantranscriptomesconvert: convert graph and alignment formatscombine: combine graphschunk: extract or break into subgraphsids: node ID manipulationsim: simulate reads by walking paths in a graphprune: prune graphs to restrict their path complexitysnarls: find bubble-like motifs in a graphmod: various graph transformationsfilter: filter reads out of an alignmentdeconstruct: create a VCF from variation in a graphpaths: traverse paths in a graphstats: metrics describing graph properties
5 文献引用
- when using
vg
https://doi.org/10.1038/nbt.4227
- when using
vg giraffe
https://doi.org/10.1126/science.abg8871
- when SV genotyping with
vg call
https://doi.org/10.1186/s13059-020-1941-7
- when using
GBZ
https://doi.org/10.1093/bioinformatics/btac656
- when using
vg deconstruct
https://doi.org/10.1038/s41586-023-05896-x
- when using
vg snarls
https://doi.org/10.1089/cmb.2017.0251
- when using
vg haplotypesand/orvg giraffe --haplotype-name
https://doi.org/10.1101/2023.12.13.571553
6 详细Wiki
6.1 安装vg
Linux编译的话推荐gcc-4.9及以上的版本。
- 安装依赖
1 | |
- 编译vg
1 | |
- Docker安装
1 | |
6.2 vg相关的格式
6.2.1 格式总览
| 名称 | 描述 | 扩展名 | 状态 | 备注 |
| VG Protobuf | 原始 vg 图格式 | .vg | 有条件使用 | 也可用于存储路径(路径不关联节点与边)。vg v1.40.0 中 vg construct 的默认输出格式(支持增量生成)。可通过 cat 拼接。通常为块 GZIP 压缩,但旧文件可能未压缩。格式为计数前缀的组,每组包含长度前缀的 Protobuf 消息,首个消息标记类型。 |
| GBZ | “GBZ”图格式,通过样本单体型路径遍历压缩存储图 | .gbz | 推荐 | 整合存储图和海量单体型数据(无需额外 GBWT 文件)。内部由 GBWT 和 GBWTGraph 组成。无法存储未被单体型路径覆盖的边。 |
| GFA | 图形化片段组装(Graphical Fragment Assembly):基于文本的图及路径存储格式。 | .gfa | 推荐(交换用) | vg 仅支持 GFA 1.x,不兼容 GFA 2。 |
| HashGraph | 基于哈希表的图格式(来自 libbdsg) | .hg, .vg | 推荐 | vg v1.40.0 多数子命令的默认输出格式。 |
| PackedGraph | 基于简洁数据结构的图格式(来自 libbdsg) | .pg, .vg | 推荐(大图适用) | 比 HashGraph 更节省空间,但速度较慢且复杂。 |
| Memory-Mapped PackedGraph | 支持增量磁盘读取的 PackedGraph 版本 | .mpg? | 试验性 | 可能未实际采用(GBZ 解决更重要的核心问题)。 |
| ODGI (vg flavor) | “优化动态基因组/图实现”(ODGI)格式的 vg 兼容版本。 | .odgi | 已移除 | 旧版 libbdsg 实现的格式与 odgi 项目不兼容,故被移除。 |
| XG | 压缩的不可变图格式(名称无实际含义)。 | .xg | 有条件使用 | PackedGraph 更优,但部分工具仍依赖旧版 XG。 |
| GBWTGraph | 将 GBWT 转译为图的补充文件(需结合 GBWT 文件使用)。 | .gg | 已弃用 | 仅存节点序列,需配合单体型 GBWT 文件使用。 |
| VG JSON | VG Protobuf 的 JSON 版本(Protobuf 图对象转为 JSON)。 | .json | 有条件使用 | 适合通过 jq 分析小图,或导入不支持 libbdsg/libvgio 的工具。一般推荐用 GFA 替代。 |
| Indexed VG Protobuf | 带索引的有序 VG Protobuf 格式(支持随机访问)。 | .sorted.vg | 已弃用 | 从未普及,已被 Memory-Mapped PackedGraph 取代。 |
| FASTA | 存储 DNA 序列的 FASTA 格式 | .fa, .fasta, .fna | 推荐(线性参考) | vg 可将其构造为线性参考基因组。 |
vg的很多文件格式都不能直接查看,需要使用vg view等才能查看;还有一些为0或者是没有设置的值夜不会展示。
6.2.2 格式框架
在vg中,.vg表示graph,.gam表示reads alignment的结果。对graph,每个message是一部分graph;对reads,每一条message是一个alignment.
6.2.3 .vg格式
.vg格式是HashGraph、PackedGraph或Protobuf graph 三种graph的格式。这三种格式的graph都可以被vg调用。现在.vg格式用的较多的是HashGraph或PackedGraph. 可以使用vg view查看GFA格式文件,使用graphviz查看pdf中的graph. 此外,也可以使用vg mod来编辑graph,但是通常的操作是先建立索引再进行操作。
6.2.4 graph术语
reference graph:表示含有参考基因组和相对于参考基因组的变异信息的graph,一个FASTA文件+VCF文件。flat graph:只包含参考基因组的graph. 因为没有bubbles,所以叫flat.augmented graph:一个graph被reads mapping以后得到的graph.augmented graph是vg genotype的基础。sample graph:含有一个或多个样品的variation的graph. 例如几个组装的基因组构建的graph,或者是单样品的reads augmented的flat graph.
6.2.5 .xg (XG lightweight graph / path index)
XG可以理解为用于构建索引的graph的轻量版。.xg包含graph的paths、nodes和edges,但是不包含对应的序列,也就是说graph的结构被保留下来,但是真实的序列被删除了。这种格式和原始格式之间的转换是很复杂的,但是使用时只需要记住:这种模式是用来查询用的,比如两个nodes之间的距离、获取相邻的两个nodes、计算nodes的degree,而所有的这些仅仅只是用到了原始graph的索引。.xg格式的索引可以用来mapping reads,也可以加速某些功能模块,这样就不用在原始的graph上进行操作。
6.2.6 .gcsa (GCSA generalized compressed suffix array index)
GCSA/GCSA2索引和bwq index生成的.sa文件是一样的。这个文件里面包含一个后缀数组,可以快速查询特定序列在graph中的位置。我们可以使用这种方法将reads mapping到graph上,这时候就可以看到GSCA索引的出现。
6.2.7 .gam(vg的BAM文件)
.gam就是vg的BAM文件,包含了某个read mapping到graph上的相对位置。这种格式支持双末端的reads. 可以使用vg view查看。这种格式也可以转换为JSON格式:
- 一行一个记录,可以是一个read,是一个JSON对象;
- 当一个值是
0/NULL/false时不存储在JSON文件中。
6.2.8 .gam.index (GAM 索引)
GAM问价有个问题就是没有隐含的序列顺序。可以使用GAM索引来查询reads,这样就不用遍历整个GAM文件。也就是说当我们想提取mapping到特定位置的所有reads或者根据位置来排序reads,那就可以用GAM索引。
GAM索引的本质是一个RocksDB数据库。这样的好处是不用讲GAM文件放入到内存中,而是存在磁盘上,这样的一个坏处是可能速度较慢。
6.2.9 .dot(DOT format for graphtools viz)
DOT是graph可视化工具graphtools的标准格式,可以直观地读取,但是这种文件最大的用处是作为vg和PDF或SVG的中间文件。
6.2.10 GFA(Graph Fragment / Assembly format)
GFA可以方便graph数据在不同的程序之间来回交换,包括比对工具、泛基因组工具和可视化工具等。应该说GFA就是构建graph-based工具的关键。另外一个优势是GFA是可读的。
6.2.11 vg扩展到GFA
vg能够调用GFA和RGFA文件,vg也能支持一些非标准的GFA标准。其中一个就是GFA头部的RS标签,vg将这个解释为空格分隔的参考样本。
6.2.12 矢量化
使用vg vectorize构建用于机器学习的向量化输出。
6.3 基础用法
6.3.1 用法总览

1 | |
6.3.2 构建泛基因组
在vg中构建泛基因组通常有两种方法,如果有参考基因组,直接使用vg construct即可。可以只使用一个参考基因组构建graph,这样构建的graph是没有bubbles,因为是自己比对到自己,所以不会产生bubbles.
1 | |
但如果只是构建这样的一个graph,那没啥意思,bwa也可以完成这个工作。真正需要的是利用一些先验信息(例如VCF文件中的变异信息)来mapping目的reads,这样做的目的是能够获得更好的mapping结果同时重新发现一些已知的变异。如果有一个使用了这个基因组得到的VCF文件,那么就可以构建这样的graph. 构建之前需要先压缩VCF文件并构建索引:
1 | |
然后开售构建graph:
1 | |
需要注意的是这一步同样需要构建参考基因组的索引,如果没有索引文件的话vg会自动创建。
可以使用-R参数指定对哪条染色体进行graph构建。理论上来说只要是 samtools-format region specifier都可以,但是通常是针对整条染色体进行构建。下面的代码是对人类基因组的染色体分批进行graph构建:
1 | |
6.3.3 vg msga替换vg construct
如果没有参考基因组,但是有polished的long reads,这时可以考虑使用vg msga进行逐步组装并构建graph:
1 | |
具体实现的步骤如下:
- 使用第一条read构建一个
flat graph - 将第二条read比对到这个graph上
- 合并新的paths
- 对graph重新构建索引并重复上两步步骤
6.3.4 构建graph索引
对泛基因组的graph构建索引和线性基因组构建索引类似,之所以构建索引是为了在后续的mapping等过程中更有效地查找泛基因组中的相关序列。
6.3.4.1 xg索引
xg格式的索引能够存储一个graph中的nodes、edges和paths. 构建完索引后就可以将graph加载到内存中进行mapping等处理,可以使用更少的内存和更少的时间,也可以更有效地查询graph中的paths. 构建的命令如下:
1 | |
6.3.4.2 GCSA2索引
GCSA2是Generalized Compressed Suffix Array的标准,可以理解为graph BWT index,类似于bwa生成的索引。可以很快速地查询特定序列在graph中的位置。例如,如果我们想查看GATTACA这个序列是否存在于所构建的graph中,这时使用GCSA2索引就可以查询到这个序列在graph中的位置。构建GCSA2索引时需要一个基于pruned de Bruijn graph的kmer参数。可以使用下面的命令构建GCSA2索引:
1 | |
运行这个代码会生成两个文件:graph.gcsa和graph.gcsa.lcp. 这两个文件必须都存在,才能保证后续使用时不报错。
针对大型且复杂的graph,例如1000个基因组构建的全基因组graph,就不可能使用所有的kmers来构建GCSA2索引。这种情况就需要在构建索引前先对graph进行修剪,保证每个复杂区域只有一个non-combinatorially-exploding subgraph.
1 | |
更多的使用方法参考:
https://github.com/vgteam/vg/wiki/Working-with-a-whole-genome-variation-graph
6.3.4.3 GBWT索引
如果输入的VCF文件包含单倍型信息,那么可以将单倍型信息加入到GBWT索引中,以获得更多的信息。GBWT能够存储大量的单倍型信息。构建索引的方法类似于XG索引。GBET索引需要XG索引的信息,所以这两中格式的索引必须同步生成。
1 | |
如果有了GBWT索引,那就可以对单倍型进行修剪处理来构建GCSA索引:
1 | |
6.3.5 Mapping reads
vg的mapping算法是MEM(maximal exact match),对应的模块是vg map. 不同的输入数据使用不同的命令,需要xg和GCSA这两种格式的索引,如果有GBWT索引的话也可以加入到其中,命令为--gbwt-name graph.gbwt.
1 | |
6.3.6 Calling variants
6.3.6.1 vg call
vg call依赖于samtools pileup格式结构,是vg中推荐的方法。实现这个方法的本质是先对graph进行增强,再使用augmented的graph来生成变异图谱。
1 | |
6.3.6.2 vg genotype
vg genotype使用类似FreeBayes的方法,但是得到的结果并不是很准确。第一步先对GAM文件构建索引,然后再识别鉴定变异。
1 | |
生成的VCF文件是未排序的,需要使用vcflib对其进行排序。
7 Mapping long reads with Giraffe
使用vg giraffe进行长reads比对。
7.1 构建索引
虽然vg giraffe会自动构建索引,但是还是建议预先构建对应的索引文件:
1 | |
会生成这样一些文件:
1 | |
.dist:距离索引.gbz:gbz格式的索引.longread.withzip.min:the “minimizers” used to find seeds, with embedded “zipcodes”.longread.zipcodes:the zipcodes too large to store in the minimizer file
7.2 Mapping
针对不同测序方法得到的数据,使用参数-b指定测序数据类型,PacBio HiFi的数据使用hifi,Oxford Nanopore R10 chemistry reads使用r10;-Z参数指定.gbz格式的graph文件;-f参数指定输入的数据类型为FASTQ;使用参数-p来展示运行过程中的信息;使用>将比对的结果输出为.gam格式。
1 | |
Giraffe能够从.gbz格式的graph中准确找到长reads的索引信息,如果没有相关的索引就会自动创建对应的索引。另外一个方法是直接把上一步生成的索引文件以传参的方式传入: --minimizer-name/-m、--zipcode-name/-z和--dist-name/-d.
Giraffe会自动预测使用多少线程来mapping,当然也可以使用-t参数指定mapping时所用的线程数。
7.3 Mapping其他格式
如果想要输出的格式为GAF格式,就使用-o参数进行指定:
1 | |
如果想要输出线性的BAM文件,那么可以使用-P进行指定:
1 | |
7.4 处理Long Read Alignments
7.4.1 描述性统计信息
1 | |
类似的输出结果:
1 | |
如果Mapping quality小于60的序列很多,那么就说明存在问题,需要检查序列质量。
7.4.2 格式转换
格式转换的时候需要graph文件。
7.4.2.1 GAM转GAF
1 | |
7.4.2.2 GAF转GAM
1 | |
7.4.2.3 剥离GAM元数据
如果想用旧版本的vg或者是其他的非vg工具查看GAM文件的话,可能会得到下面的信息:
1 | |
处理方法是将GAM转为GAF再转为GAM,这样就会把这些元数据去掉了。
7.4.3 将长reads添加到BAM
在有GMA或者是GAF格式文件后,如果想得到线性参考的BAM文件用于DeepVariant等软件,那么可以使用vg surject来surject比对结果并压缩为线性参考。对于长reads,在使用vg surject时需要添加-prune-low-cplx/-p参数,这样能够更好地处理跨越了串联重复等区域的reads.
1 | |
如果是GAF文件,对应的命令为:
1 | |
7.5 最佳实践操作
短reads同样适用。
- 双末端mapping(合二为一)
1 | |
- 双末端mapping
1 | |
- 双末端mapping后输出BAM文件
1 | |
8 单倍型sampling
泛基因组graph作为参考进行比对时,嵌入到graph中的variation会影响mapping的结果。
- 在graph和reads中都存在的variant会使mapping更准;
- variant只存在于graph中但是不存在于reads中的话,mapping会很慢,准确度也不高。
常见的是只使用共有的/常见的variant,但是后面开发出一个新的方法:对每个样品都构建一个个性化的reference. 这种方法是通过计算reads中的kmers,并根据kmer的数量来生成一个小的合成的单倍型。
8.1 文献
https://www.nature.com/articles/s41592-024-02407-2
8.2 处理graph
1 | |
假设graph是由多个样本alignment后构建的,有着很高水平的线性结构,例如Minigraph-Cactus pipeline构建的graph。graph和单倍型必须是GBZ格式。预处理方法是将每条染色体切成长度为10 kbp的短序列,然后在其中选择最相关的单倍型,并将这些单倍型连接形成临时的合成单倍型;同时需要选择一些特定的kmers,通过这些kmers的存在和缺失来表示单倍型。