vi ~/mambaforge/envs/metawrap-env/bin/config-metawrap # Paths to metaWRAP scripts (dont have to modify) mw_path=$(which metawrap) bin_path=${mw_path%/*} SOFT=${bin_path}/metawrap-scripts PIPES=${bin_path}/metawrap-modules
# CONFIGURABLE PATHS FOR DATABASES (see 'Databases' section of metaWRAP README for details) # path to kraken standard database # KRAKEN_DB=~/KRAKEN_DB KRAKEN2_DB=~/database/kraken2.plus.plantandfungi
# path to indexed human (or other host) genome (see metaWRAP website for guide). This includes .bitmask and .srprism files BMTAGGER_DB=~/database/human.genome # # paths to BLAST databases BLASTDB=~/database/ncbi.nt TAXDUMP=~/database/ncbi.taxonomy
for F in 0.data/*_1.fastq; do R=${F%_*}_2.fastq; BASE=${F##*/}; SAMPLE=${BASE%_*}; metawrap read_qc --skip-bmtagger -1 $F -2 $R -t 70 -o 1.read.qc/$SAMPLE & done
将质控后的数据移动到新的文件中:
1 2 3 4 5 6
mkdir -p 2.clean.reads/all for i in 1.read.qc/*; do b=${i#*/} mv${i}/final_pure_reads_1.fastq 2.clean.reads/${b}_1.fastq mv${i}/final_pure_reads_2.fastq 2.clean.reads/${b}_2.fastq done
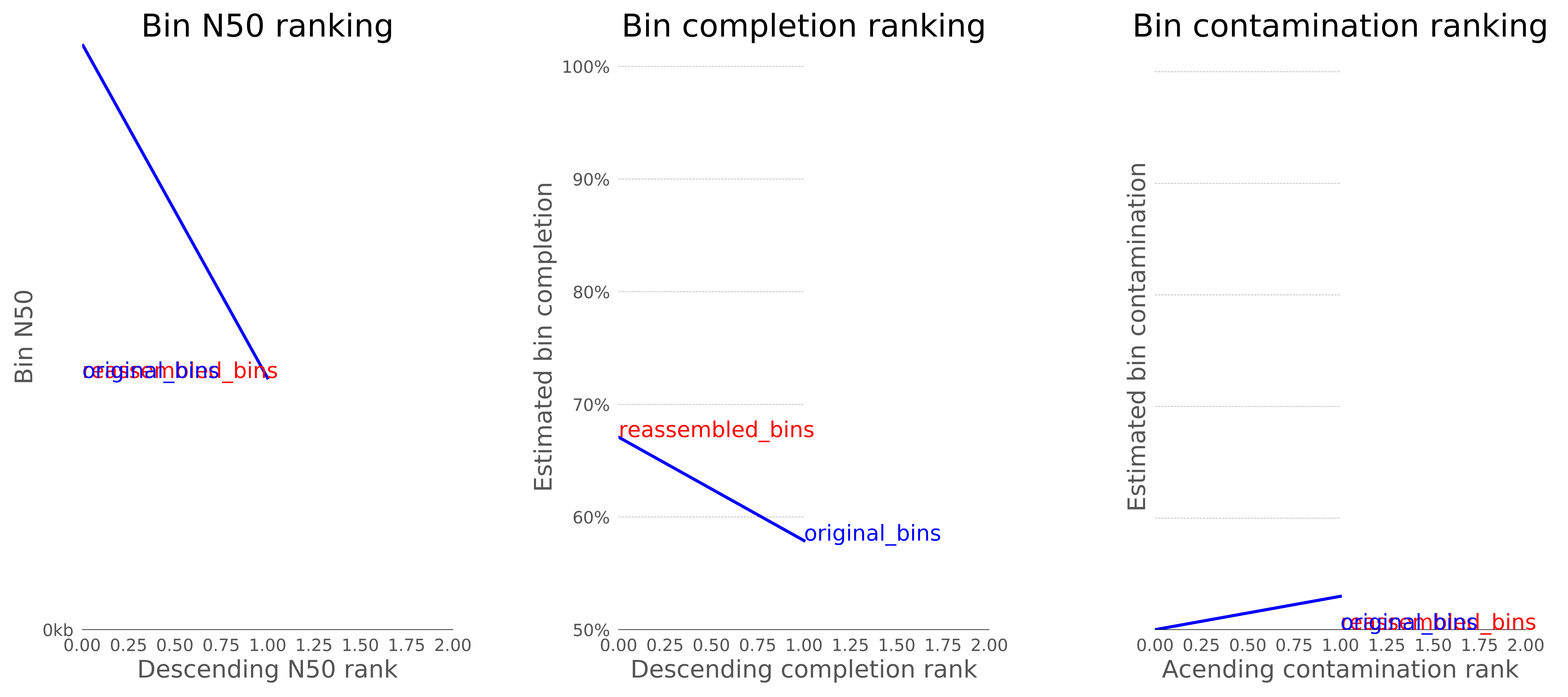
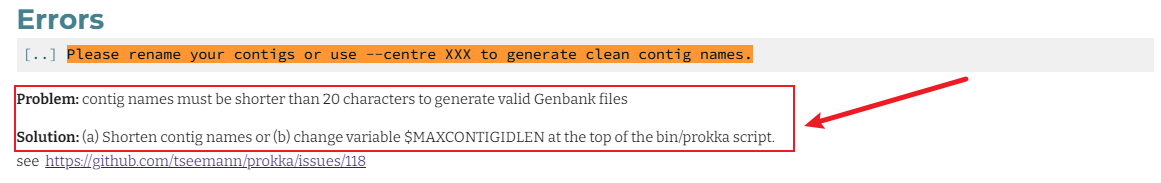
mkdir 9.bin.reassembly/renamed.bins mkdir 9.bin.reassembly/renamed.bins.id for i in 9.bin.reassembly/reassembled_bins/*; do python3 ~/scripts/rename.fasta.id.py ${i} 9.bin.reassembly/renamed.bins/${i##*/} 9.bin.reassembly/renamed.bins.id/${i##*/}.id.txt; done
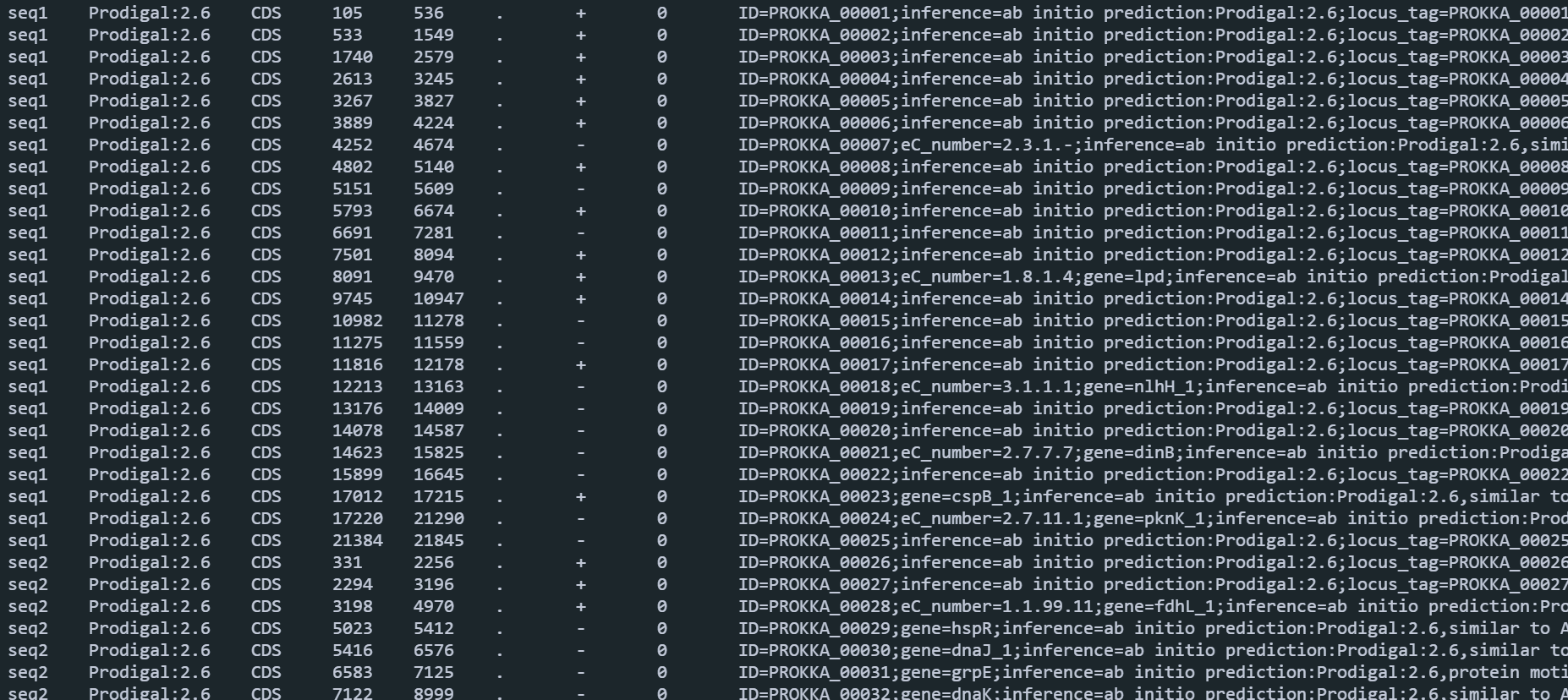
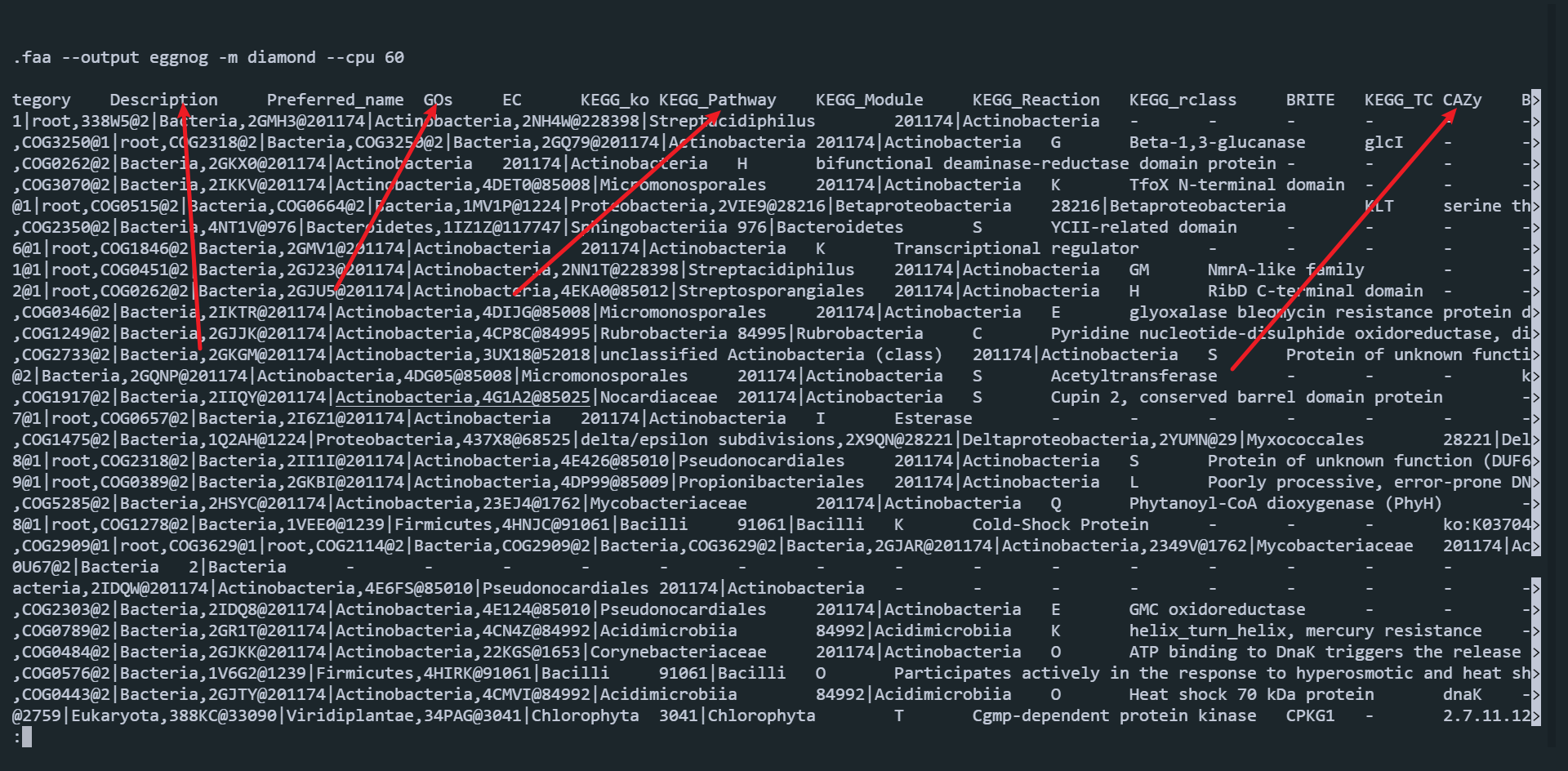
mkdir 12.eggNOG-mapper for i in 11.bins.anno/bin_translated_genes/*; do python3 ~/software/eggNOgmapper/emapper.py -m diamond --cpu 60 -i 11.bins.anno/bin_translated_genes/${i##*/} --output 12.eggNOG-mapper/${i##*/}.eggnogmapper.txt; done
for F in 0.data/*_1.fastq; do R=${F%_*}_2.fastq; BASE=${F##*/}; SAMPLE=${BASE%_*}; metawrap read_qc --skip-bmtagger -1 $F -2 $R -t 70 -o 1.read.qc/$SAMPLE & done
mkdir -p 2.clean.reads/all
for i in 1.read.qc/*; do b=${i#*/} mv${i}/final_pure_reads_1.fastq 2.clean.reads/${b}_1.fastq mv${i}/final_pure_reads_2.fastq 2.clean.reads/${b}_2.fastq done
mkdir 9.bin.reassembly/renamed.bins mkdir 9.bin.reassembly/renamed.bins.id for i in 9.bin.reassembly/reassembled_bins/*; do python3 ~/scripts/rename.fasta.id.py ${i} 9.bin.reassembly/renamed.bins/${i##*/} 9.bin.reassembly/renamed.bins.id/${i##*/}.id.txt; done
mkdir 12.eggNOG-mapper for i in 11.bins.anno/bin_translated_genes/*; do python3 ~/software/eggNOgmapper/emapper.py -m diamond --cpu 60 -i 11.bins.anno/bin_translated_genes/${i##*/} --output 12.eggNOG-mapper/${i##*/}.eggnogmapper.txt; done