1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
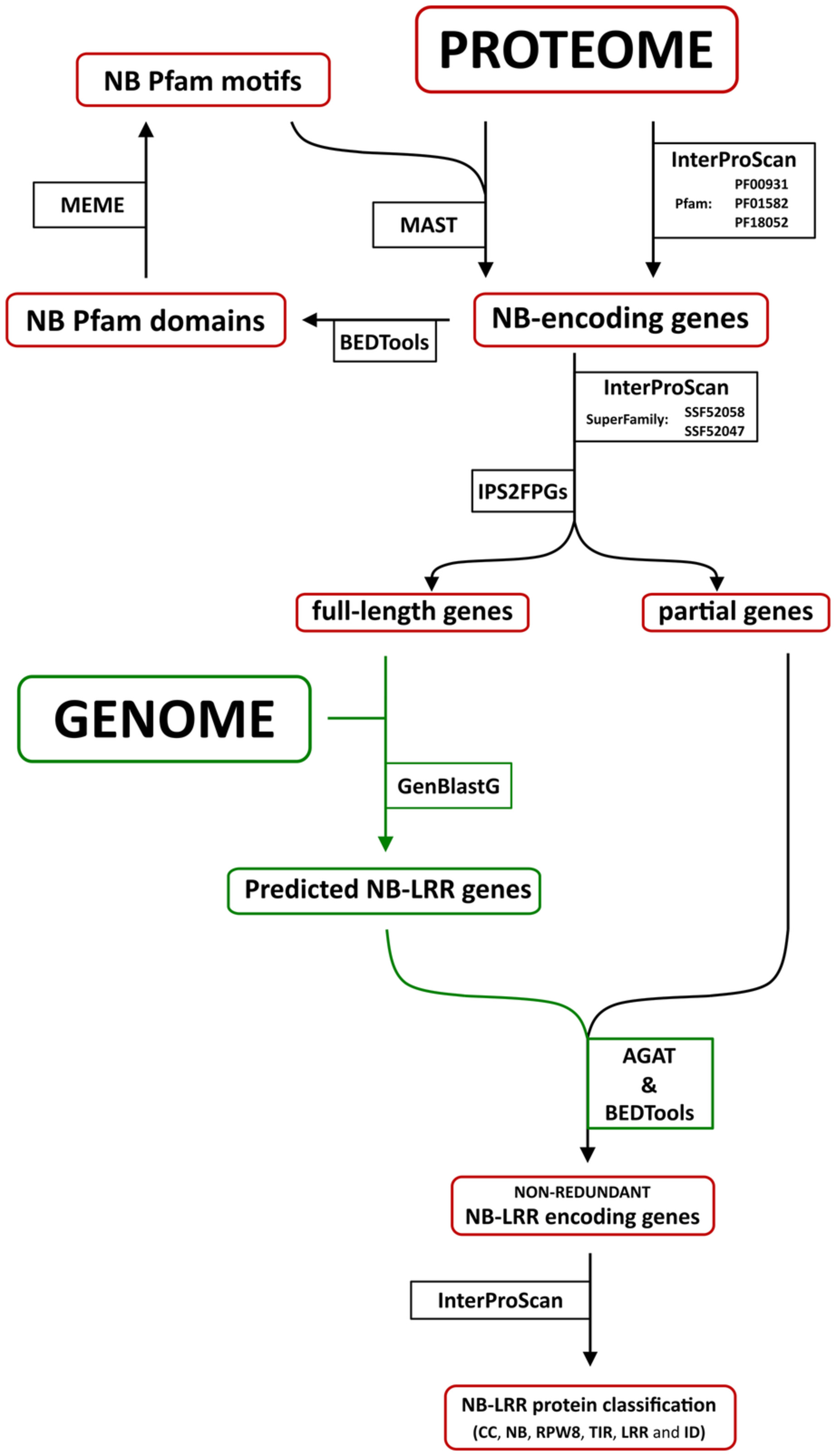
| bash /usr/bin/interproscan.sh -f TSV -app Pfam -i data/proteins.fa -b results/1-interproscan
grep NB-ARC results/1-interproscan.tsv | cut -f1,7,8 > results/2-interproscan.bed
bedtools getfasta -fi data/proteins.fa -bed results/2-interproscan.bed -fo results/3-extracted.fasta
meme results/3-extracted.fasta -protein -o results/4-meme_out -mod zoops -nmotifs 19 -minw 4 -maxw 7 -objfun classic -markov_order 0
mast -o results/5-mast_out results/4-meme_out/meme.txt data/proteins.fa
bash /usr/bin/interproscan.sh -f TSV -app SUPERFAMILY -i results/3-extracted.fasta -b results/6-lrr_domain.tsv
bash code/IPS2fpGs.sh -f -c code/conf1.tsv -d code/conf2.tsv -o results/7.1-full_or_partial.tsv results/1-interproscan.tsv
bash code/IPS2fpGs.sh -f -c code/conf1.tsv -d code/conf2.tsv -o results/7.2-full_or_partial.tsv results/6-lrr_domain
awk '{if($2 == "full-length") print $1}' results/7.1-full_or_partial.tsv > results/8-full_length.id
perl /usr/bin/get_fa_by_id.pl results/8-full_length.id data/proteins.fa results/9-full_length.fasta
/opt/software/genblast/genblastg -q results/9-full_length.fasta -t data/genome.fa -gff -cdna -pro -o results/10-genblastG_out
perl /usr/bin/agat_sp_filter_gene_by_length.pl --gff results/10-genblastG_out_1.1c_2.3_s1_0_16_1.gff --size 20000 --test "<" -o results/10-filtered.length.gff
grep transcript results/11-filtered.length.gff | gff2bed | sortBed | clusterBed -s | cut -f4,11 > results/12-clusters
awk 'BEGIN{FS="[> ]"} /^>/{val=$2;next} {print val,length($0);val=""} END{if(val!=""){print val}}' results/*.pro | tr ' ' \\t > results/13-estimation
join -t $'\t' -1 1 -2 1 -o 1.1,1.2,2.2 <( sort -bk1 results/12-clusters) <(sort -bk1 results/13-estimation) | sort -bk2,2 -bk3,3 -nr | sort -uk1,1 | cut -f1 > results/14-r_gene_id_list
Rscript code/get_unique_id.R
perl /usr/bin/get_fa_by_id.pl results/15-ubique.r.gene.id data/proteins.fa results/16-candidate.gene.fa
bash /usr/bin/interproscan.sh -f TSV,GFF3 -i results/16-candidate.gene.fa -b results/17-annotation.results
|
1
2
3
4
5
6
7
8
9
| rm(list = ls())
library(tidyverse)
data.table::fread("results/14-r_gene_id_list", header = FALSE) %>%
dplyr::mutate(id = stringr::str_sub(V1, 1, 15)) %>%
dplyr::select(id) %>%
dplyr::distinct_all() %>%
data.table::fwrite(file = "results/15-ubique.r.gene.id", row.names = FALSE, col.names = FALSE)
|
💌lixiang117423@foxmail.com
💌lixiang117423@gmail.com