ggtree学习笔记
写在前面
Y叔的ggtree $^{[1]}$ 毫无疑问是当前绘制美化系统发育树(下文简称进化树)的最佳工具,一直想学习,但是都没有真真正正学习过,一是因为网上关于gtree的中文资源较少,另外一个原因是感觉到自己用不上,就没认真学习。春节在家,实在无聊,下定决心学一遍ggtree。下面的内容来自Y叔的博客$^{[2]}$ ,若有不当之处,恳请批评指正。
关于进化树
进化树怎么看
进化树展示的是进化关系,简单说就是亲缘关系,通常是使用物种的遗传序列(如DNA序列、氨基酸序列等)来构建的。进化树看起来和层次聚类很像,这两者有木有区别呢?Y叔在统计之都上的文章$^{[3]}$ 是这样描述的:
层次聚类的侧重点在于分类,把距离近的聚在一起。而进化树的构建可以说也是一个聚类过程,但侧重点在于推测进化关系和进化距离 (evolutionary distance)。
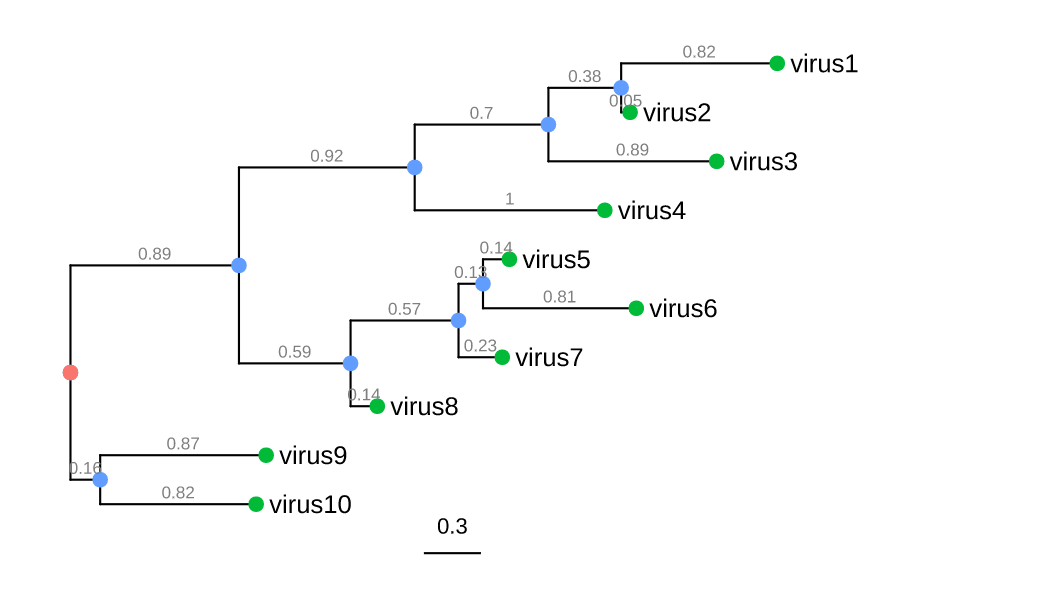
上图展示的是典型的系统发育树。图中绿色、蓝色和红色的点都是nodes,也就是节点(个人理解),其中最外层绿色的点表示的是每个sample,这时绿色的点也叫tips;蓝色的点表示的是父节点,也就是从外往内两两sample的共有节点,可以理解成祖先(ancestor),祖先之间还可以继续往上溯源,最终就汇集到红色的点,这个红色的点也就是root,需要注意的是有根树才具有root节点;横线叫做分支(branches),这些横线表示的是进化变化(evolutionary changes),线的长短表示的是以时间或遗传变异为单位的进化变化。
进化树数据格式
进化树的数据格式有多种,常见的有Newick、NEXUS及Phylip。
Newick格式
Newick格式是最常见的使用最广泛的进化树数据格式。
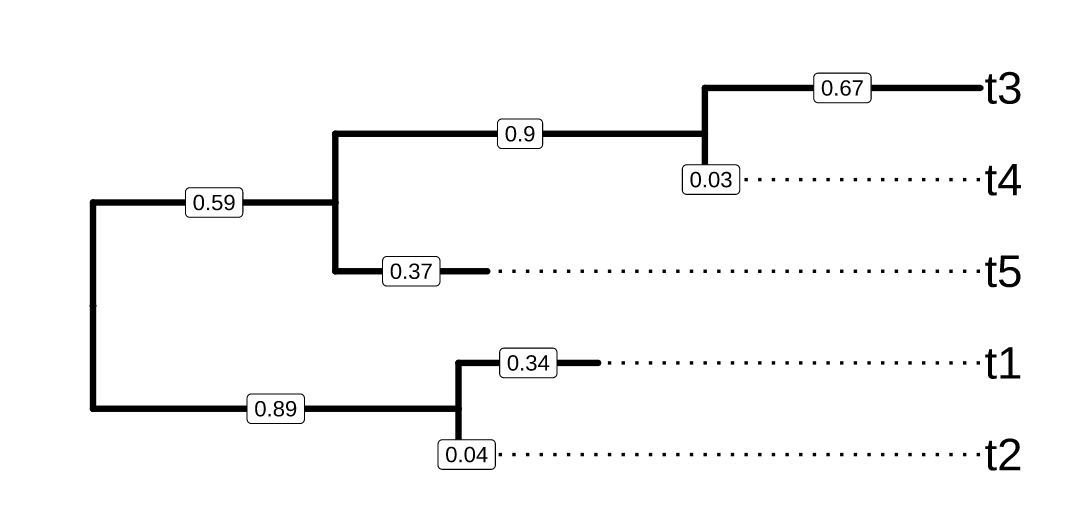
上图是一个标准的进化树图,其对应的Newick格式为:
1 | |
数据格式和图是一一对应的,假如我们看图的时候是从外到内,那t3和t4是最近的,在数据中,t3和t4也是在一个括号里面的,数据的最小单位是一对(),就像剥洋葱那样顺着括号一层一层往外剥的时候,就能得到上图的那种样式。冒号后面对应的是横线上的“距离”,父节点的“距离”是两个“子节点”共有的,因此,需要表示“父节点”的“距离”时,需要将“距离”放在“子节点“的括号外。
NEXUS格式
NEXUS格式是Newick格式的拓展,以blocks为单位将进化树的元素分开。
1 | |
其他格式
进化树的存储格式还有很多种,更多请参照Y叔的博客$^{[4]}$。
进化树数据处理
进化树数据可以使用treeio$^{[5]}$ 这个包进行合并等操作,然后可以将其他信息利用tidytree$^{[6]}$ 这个包将树文件转换成R里面常见的数据框格式,这种格式也可以再次转化成树文件,利用ggtree进行可视化。
phylo对象
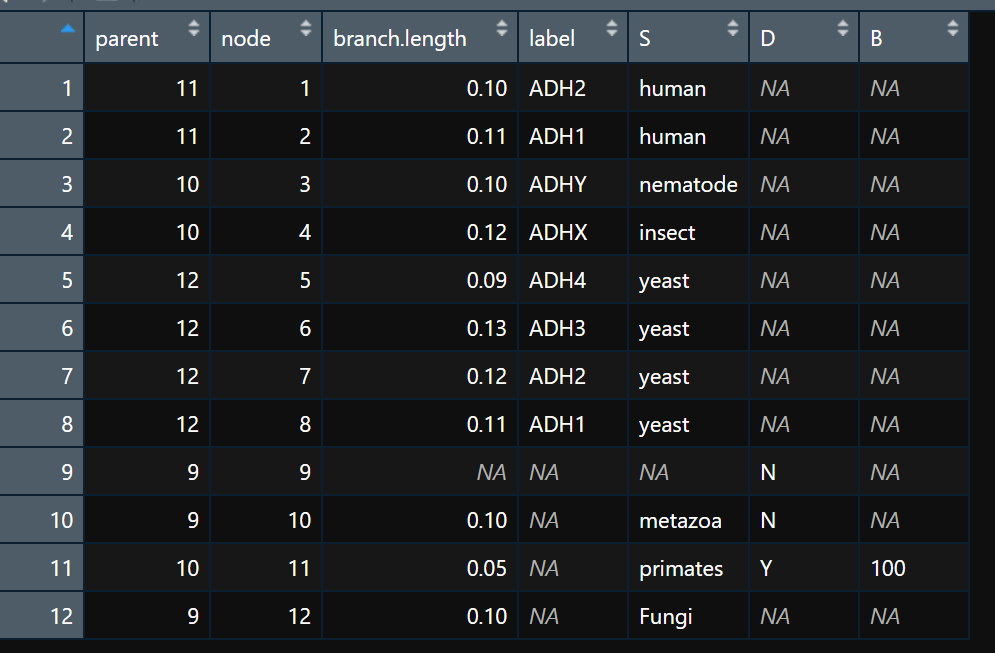
phylo格式是R包ape支持的格式,在R中的很多包都依赖于这种格式。tidytree中的函数as_tibble可以将phylo转换成数据框,此时的数据框是个tbl_tree对象。
1 | |
此时得到的是phylo对象:
1 | |
现在将其转化成数据框:
1 | |
1 | |
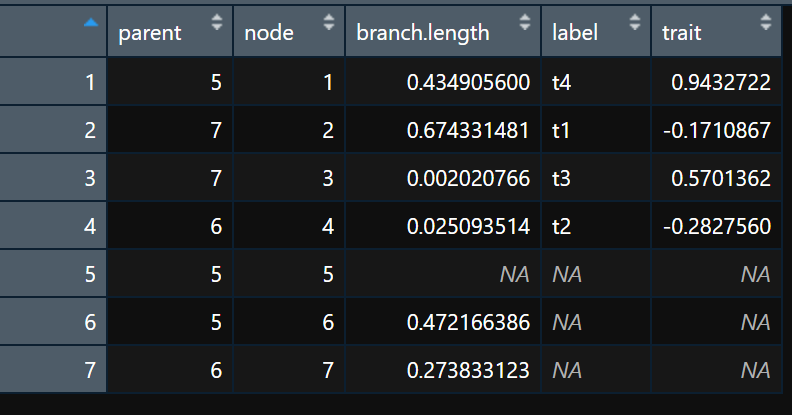
看图更直观:
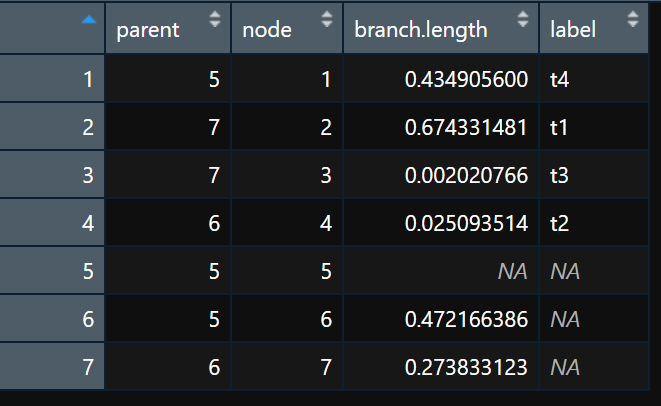
可以清楚地看到进化树的全部信息,包括父节点、节点、分支长度及tips的标签等。
使用as.phylo(x)就能将数据框转换成phylo:
1 | |
如果此时我们需要添加信息的话,在原始文件上添加信息是比较麻烦的,但是可以团购先构建数据框,然后将两个数据框join在一起就可以了:
1 | |
treedata对象
tidytree默认的格式是treedata,函数as.treedata可以将前面的数据框转换成treedata对象:
1 | |
1 | |
同样也可以通过as_tibble将treedata转换成数据框格式:
1 | |
1 | |
树文件融合
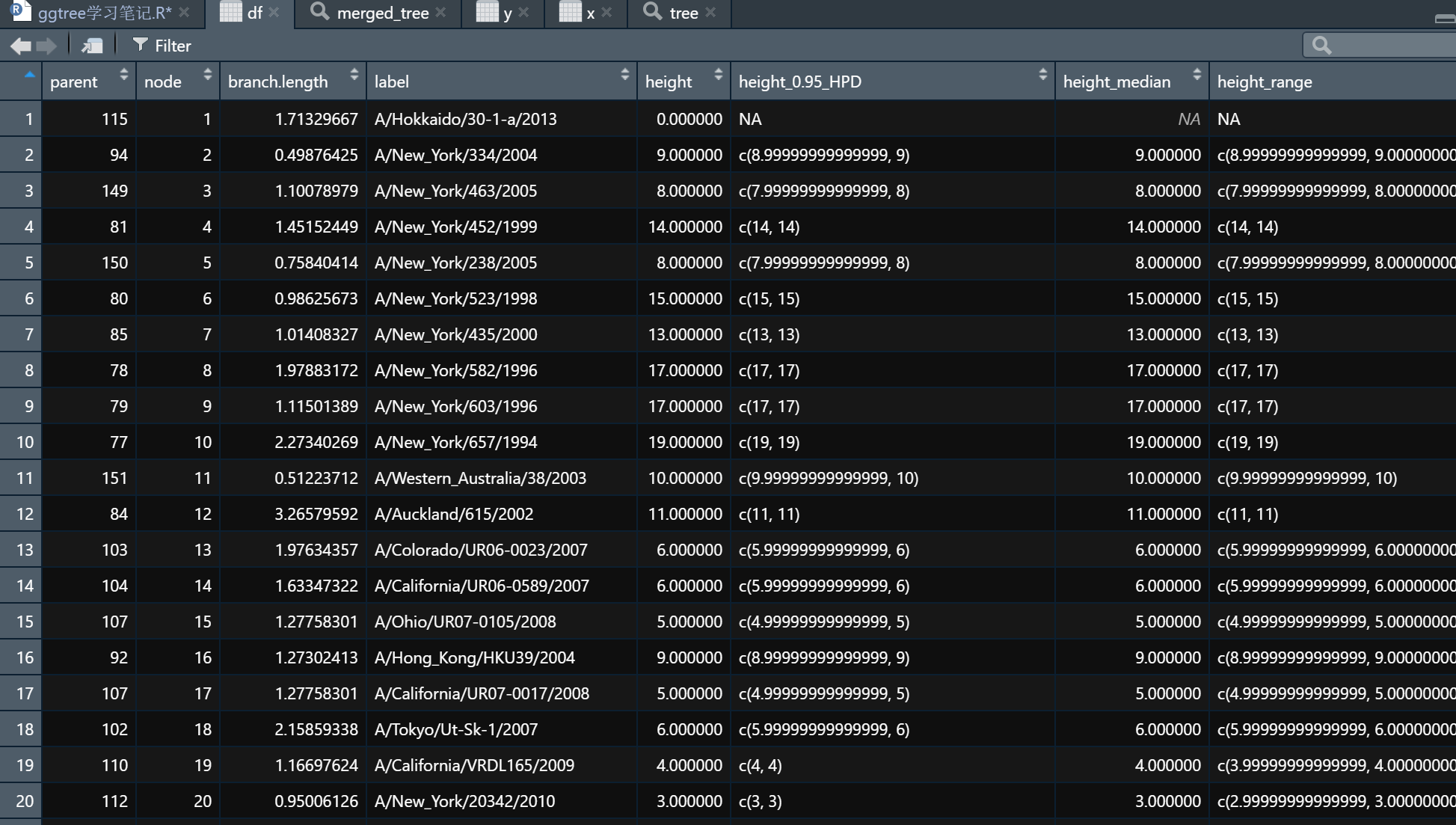
treeio同的函数merge_tree()可以对多个树文件进行合并,原理是以node/branches为`key进行数据合并,如:
1 | |
1 | |
合并后的树文件,除开node和branches外,其余的所有附加信息都变成了变量。
既然是个数据框,那就可以对这些数据进行可视化:
1 | |
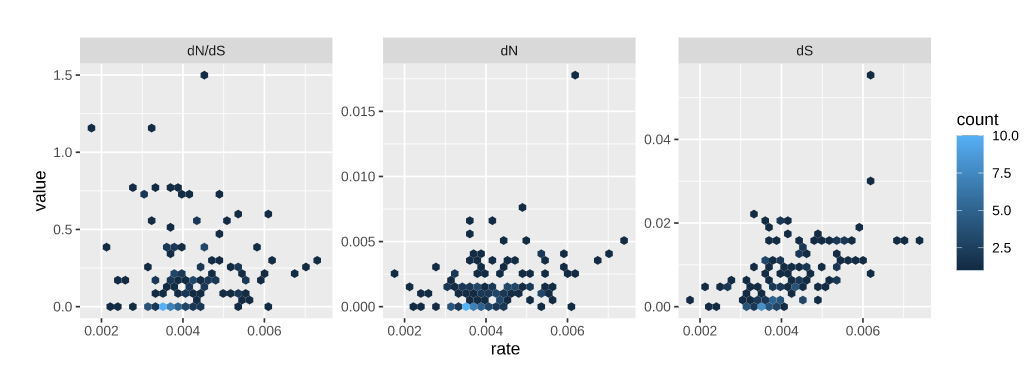
还可以利用该函数比较不同软件的分析结果:
1 | |
链接外部数据
进化树能够展示的东西不仅仅是进化关系,还可以增添许多信息,如基因表达量啥的。treeio的函数full_join()能够通过node或tips进行数据融合:
通过
node:1
2
3
4
5
6
7
8
9
10
11
12library(ape)
data(woodmouse)
d <- dist.dna(woodmouse)
tr <- nj(d)
bp <- boot.phylo(tr,
woodmouse,
function(x) nj(dist.dna(x)))
bp2 <- tibble(node=1:Nnode(tr) + # 计算父节点数
Ntip(tr), # 计算tip数
bootstrap = bp)
full_join(tr, bp2, by="node")通过
tips:1
2
3
4file <- system.file("extdata/BEAST", "beast_mcc.tree", package="treeio")
beast <- read.beast(file)
x <- tibble(label = as.phylo(beast)$tip.label, trait = rnorm(Ntip(beast)))
full_join(beast, x, by="label")
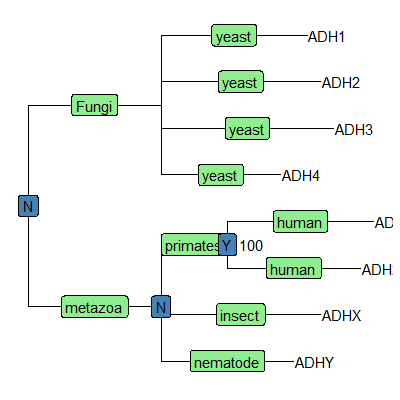
如何找到想要的node
1 | |
分组
treeio中的函数 groupOTU() 和 groupClade() 可以用于分组。
groupClade
1 | |
1 | |
以node17和node21为界将clade进行分组。这两个函数可以作用于tbl_tree、phylo 、 treedata及 ggtree 这些对象。
groupOTU
1 | |
1 | |
更常见的分组方法是直接命名分组:
1 | |
1 | |
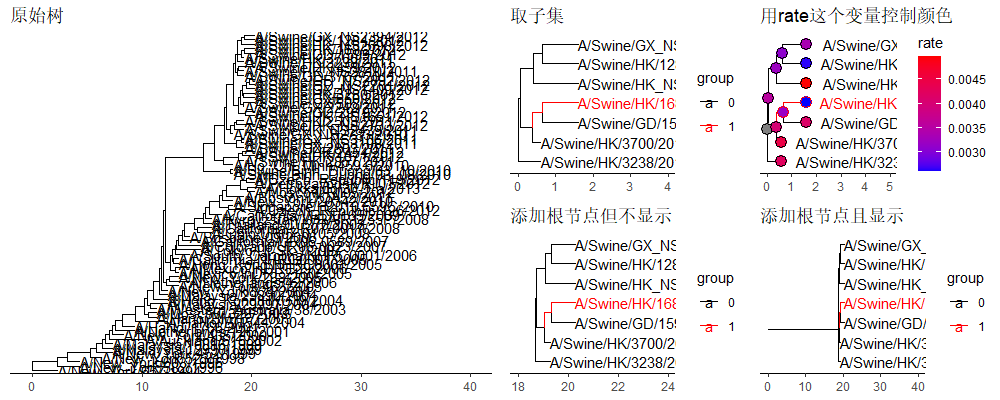
重新标准化分支
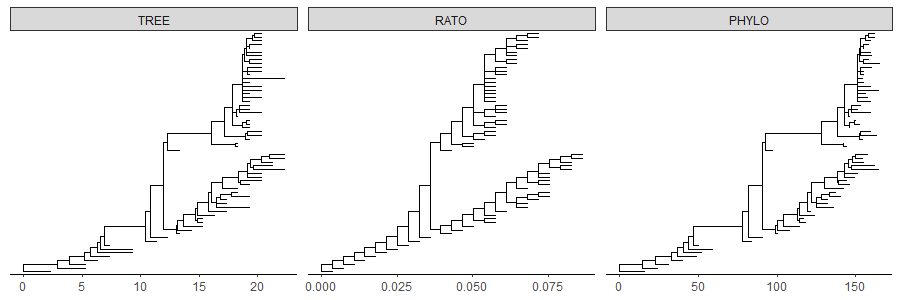
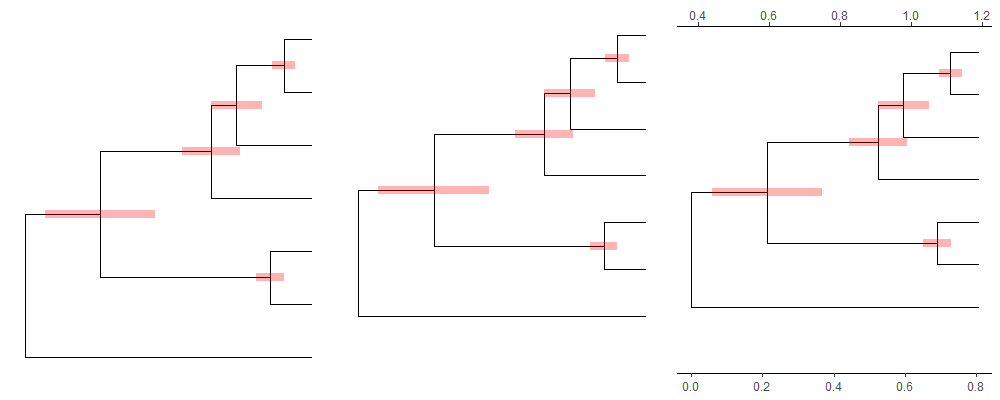
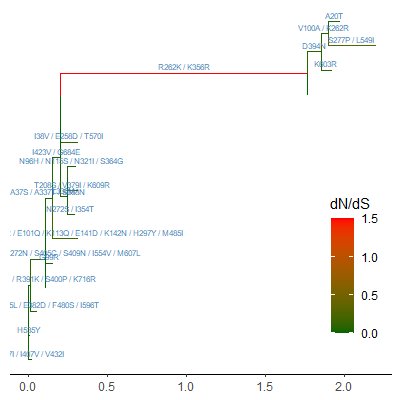
不同的进化树可以进行合并,原始的分支长度可能单位不一样,这时候就可以用合并后的其他参数对分支长度进行标准化。
1 | |
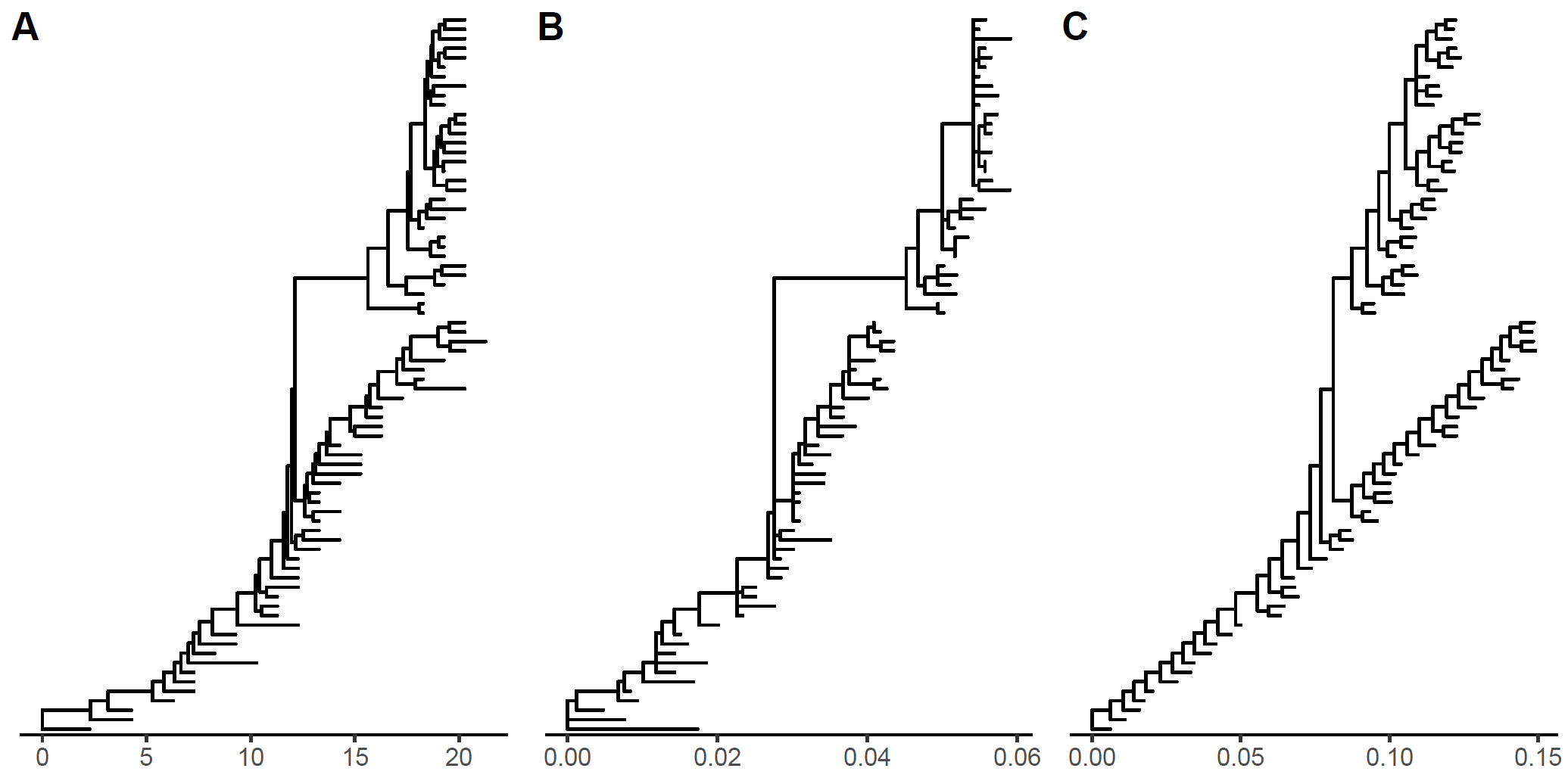
从上图可以看到的是,三个图的“横坐标”范围不一样,第一个图是原始的分支单位,第二个是以dN进行标准化的结果,第三个是以rate进行标准化的结果。
子集操作
从树中除去tips
有些时候出于某些原因(如序列质量、组装质量、比对错误等),我们会将某些tips(样本)从树里面剔除,函数drop.tip()可以实现这一功能:
1 | |
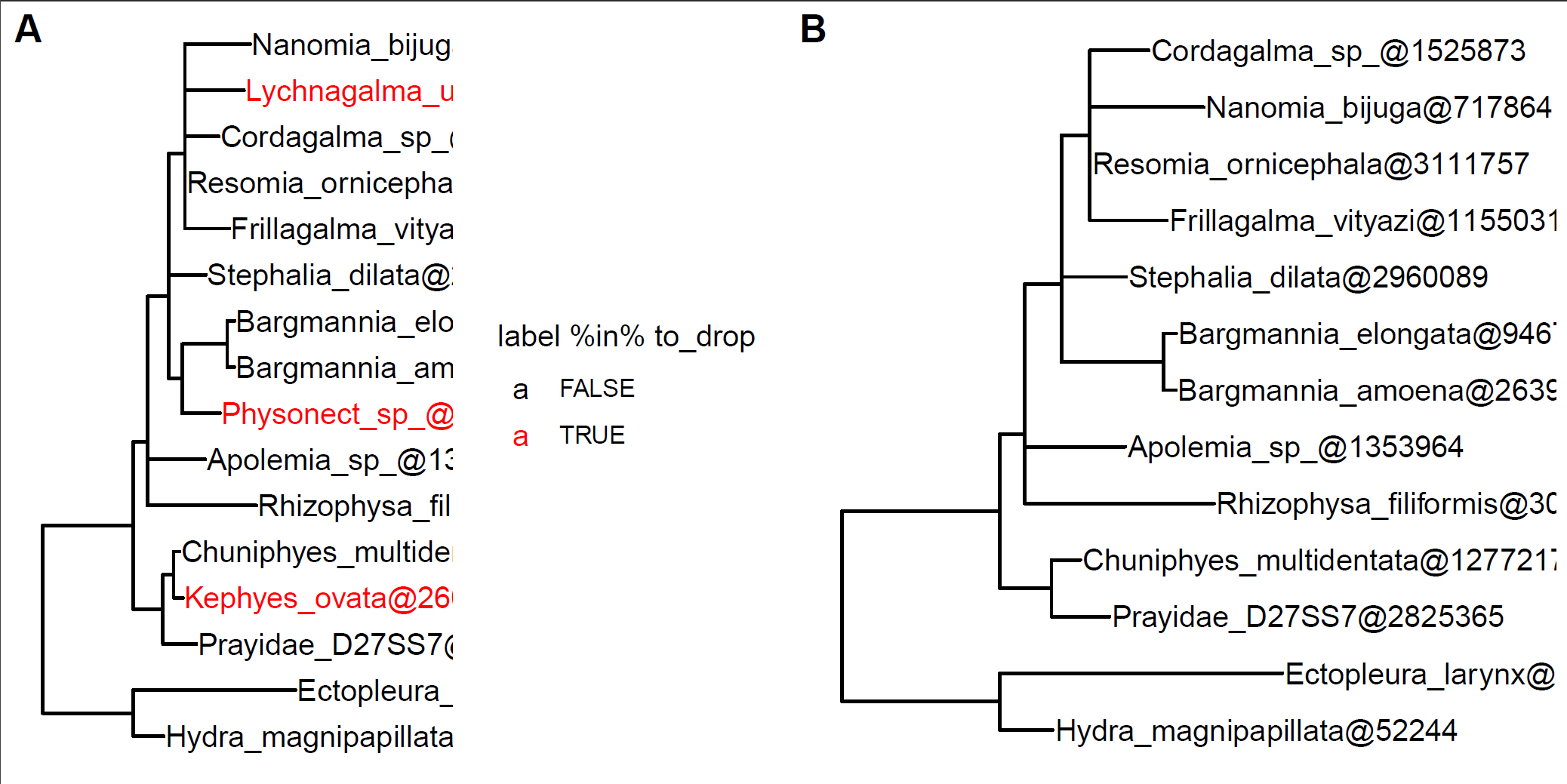
根据tips取子集
如果一个进化树很复杂的话,要看我们感兴趣的部分就很难,这时候就需要将我们感兴趣的部分提取出来。treeio中的函数tree_subset()能够完成这一功能,即使是提取出来的子集,结构还是和原来的一样,不会发生变化。
1 | |
根据内部节点编号取子集
如果我们对特定的进化分支(clade)感兴趣,那也可以通过tree_subset()函数将感兴趣的分支进行放大展示,但是这个时候需要我们知道感兴趣的进化分支所对应的node编号才行。
1 | |

导出数据
treeio这个R包可以导出多种格式的文件,这个比较简单,详情参照:https://yulab-smu.top/treedata-book/chapter3.html
进化树可视化
基础方法
ggtree对进化树进行可视化的方法有两种:
ggplot()+geom_tree()+theme_tree()ggtree()
第二种方法是第一种方法的“缩写版”。
1 | |
这两种方法得到的结果是一样的:
ggtree支持ggplot2的图形语法,因此,也可以在ggtree中对颜色性状等进行修改:
1 | |
可以使用参数branch.length对egde进行标准化,如果参数为none,则是这样:
1 | |
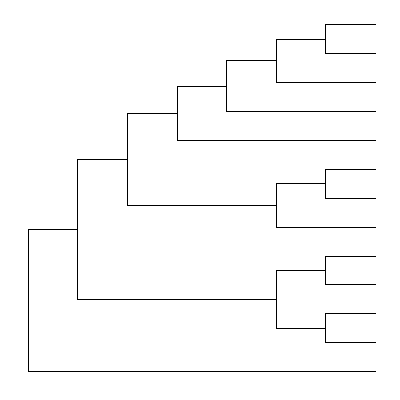
输出样式
ggtree支持多种输出样式:
1 | |

当只是展示树结构而没有分支长度标尺的时候,就用最下面这4种。
还有其他的多种对齐方式:
1 | |

如果是时间范围数据的话,则需要调用参数mrsd(most recent sample data):
1 | |
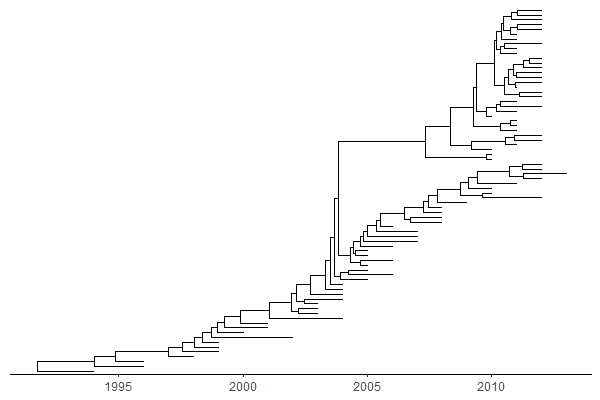
展示相关元素
展示进化距离
函数geom_treescale()用于展示进化距离。
1 | |

展示nodes/tips
1 | |
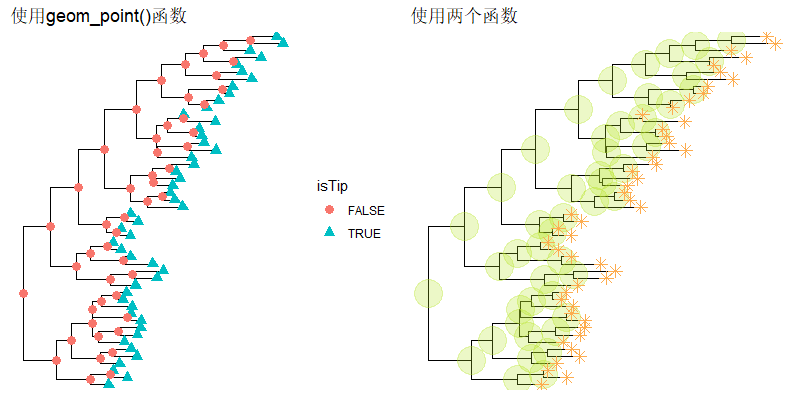
展示tips的标签
1 | |

展示根节点
1 | |
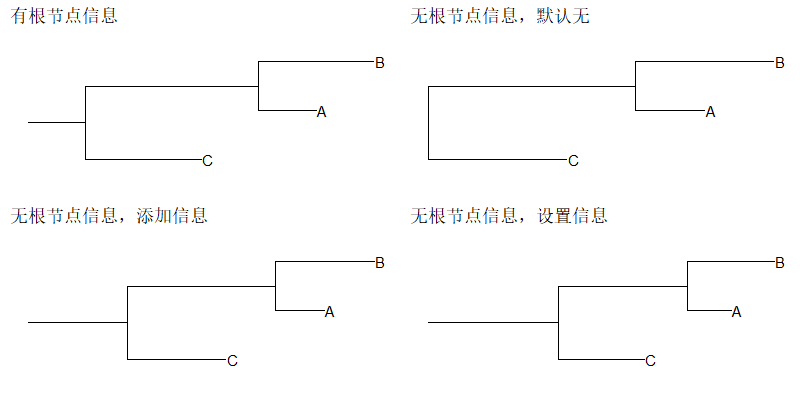
上色
上色直接是很简单,就像ggplot2那样:
1 | |
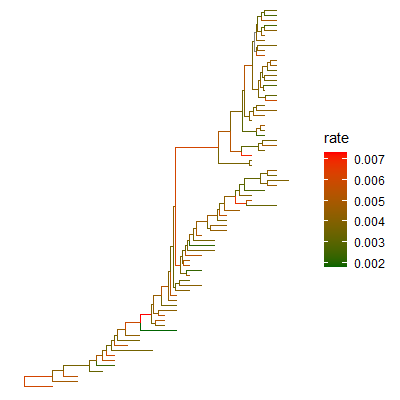
1 | |
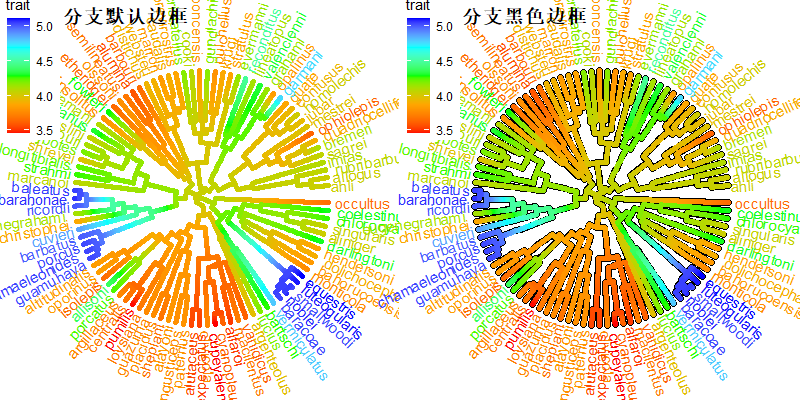
对树进行重新标准化
多个参数对进化树进行标准化,时间序列相关的参数用msrd,其余的参数可以用branch.length。
1 | |

也可以用rescale_tree对树进行标准化:
1 | |
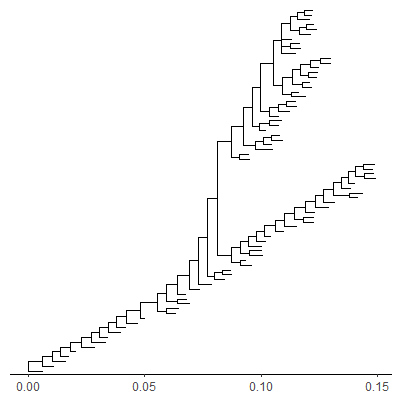
背景色
1 | |
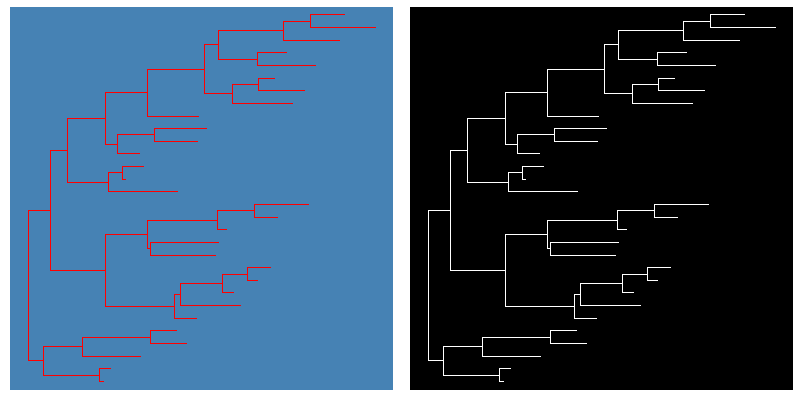
批量建树
批量建树就相当于ggplot2中的分面:
1 | |
进化树注释
基本用法
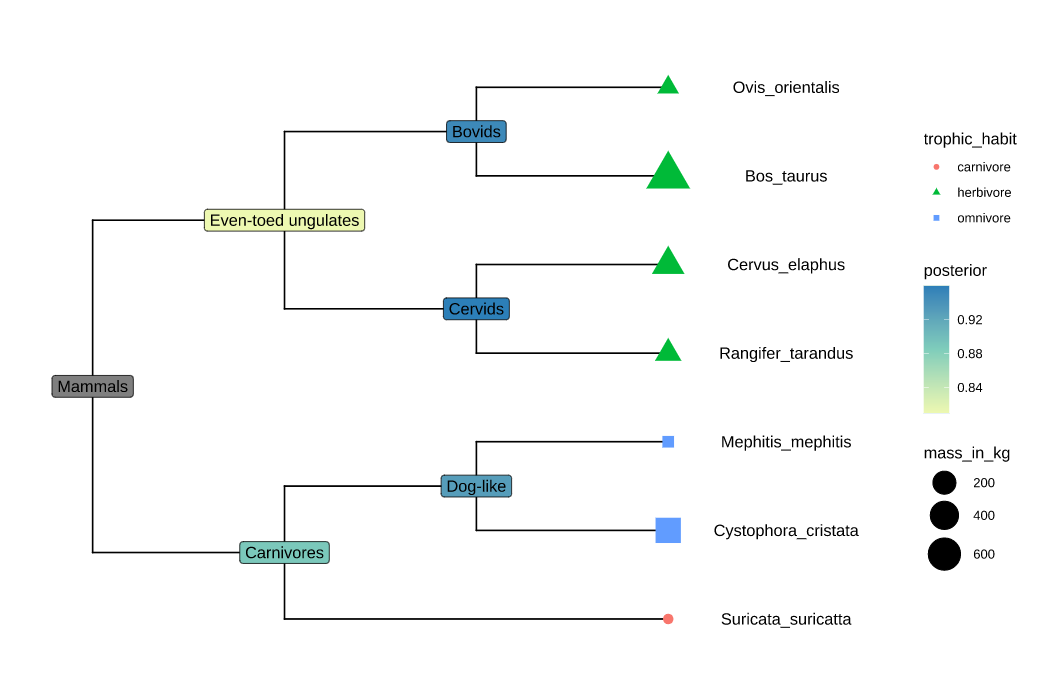
注释的信息很多,节点的分组、样品来源、基因表达量等都可以作为注释的信息。一个简单的例子:
1 | |
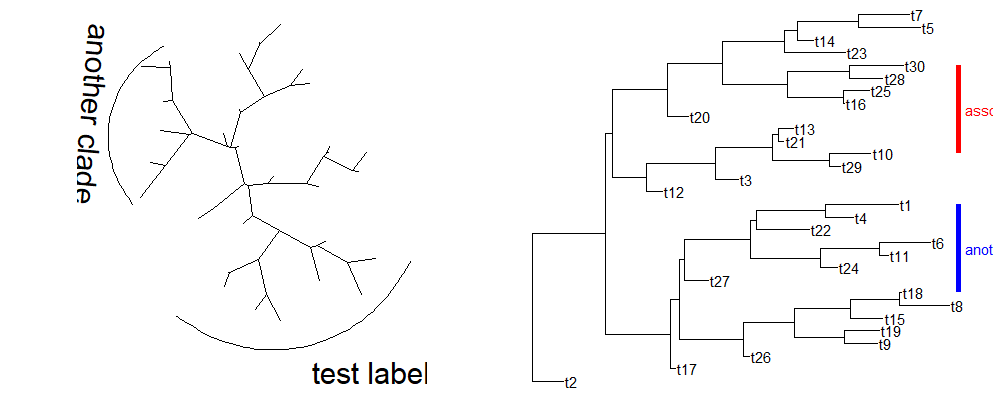
clade注释
1 | |

同样也适用于无根输出样式,可以根据node的编号,也可以直接用tips的标签(这个是真的赞啊):
1 | |
局部高亮
局部高亮可以根据node编号进行高亮,也可以使用附加数据或树数据里面的数据进行高亮:
1 | |
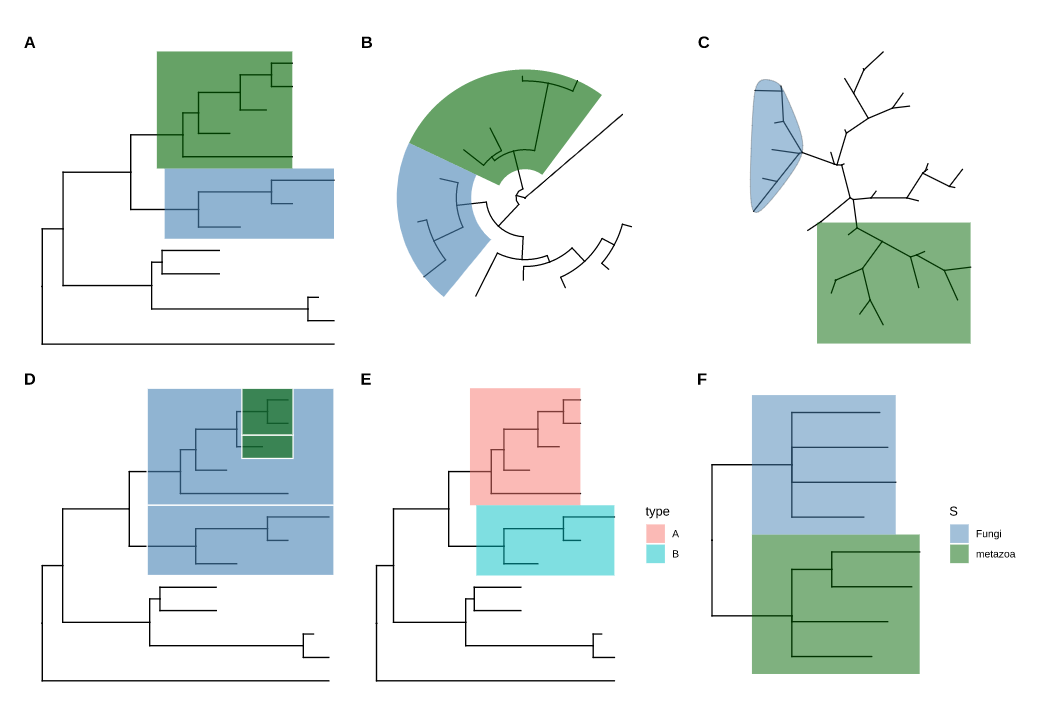
高亮不同分组
1 | |
样品连接
利用函数geom_taxalink()可以实现样品之间的连线,但是只支持有限的几种形式:
1 | |
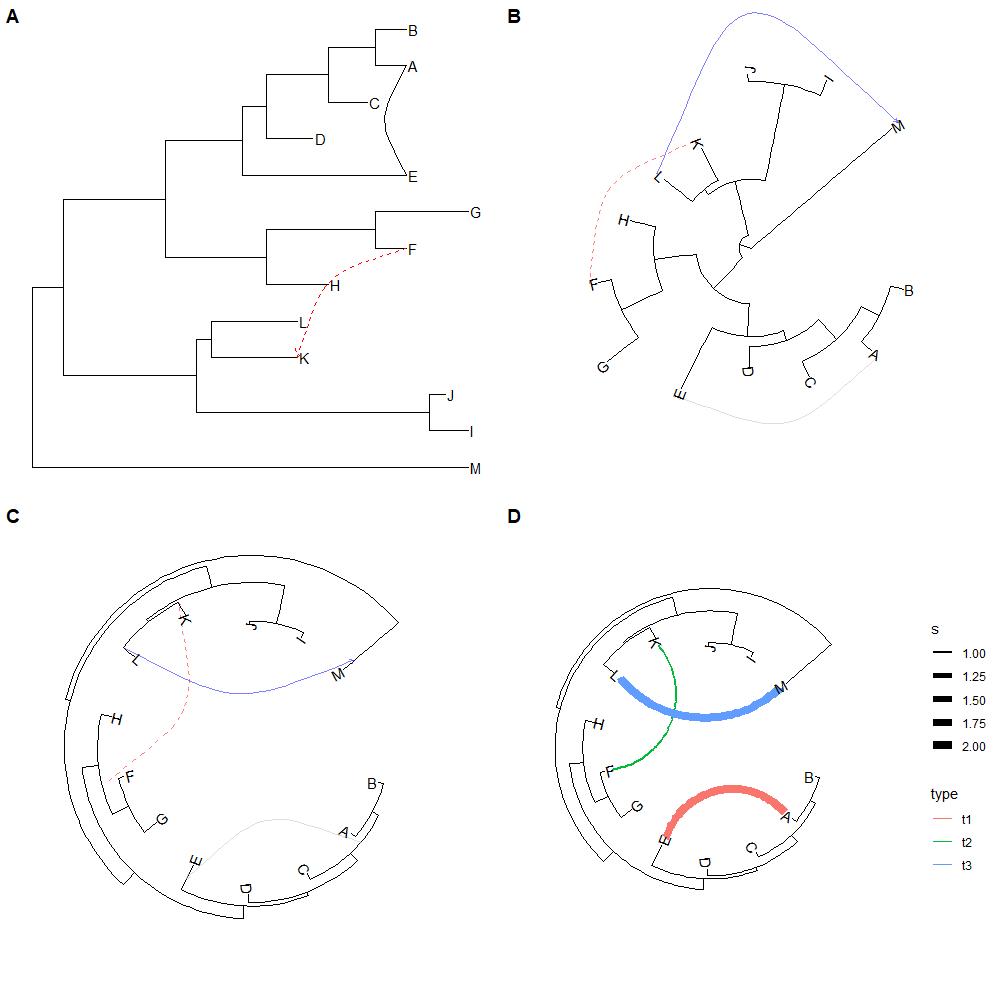
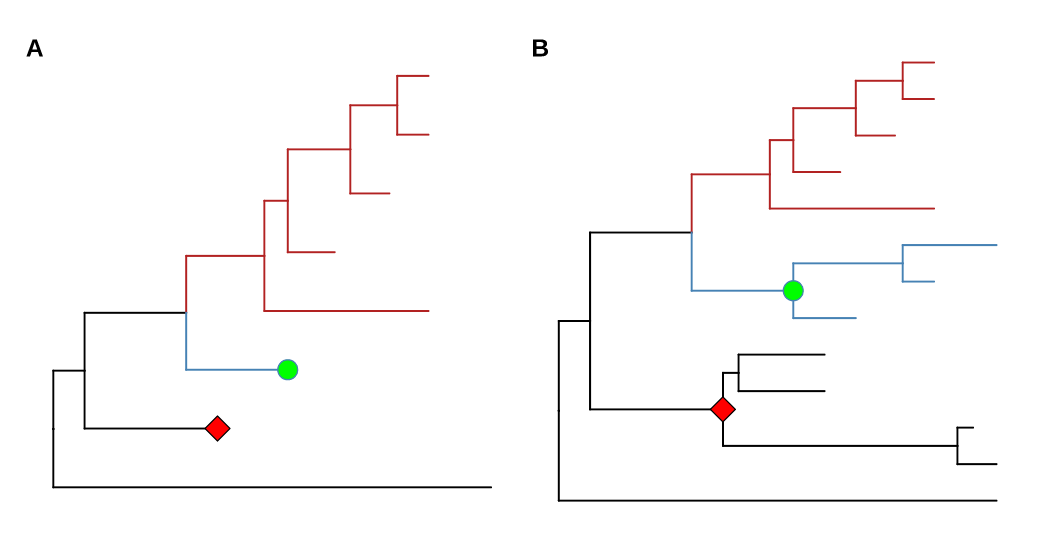
Uncertainty of evolutionary inference
1 | |
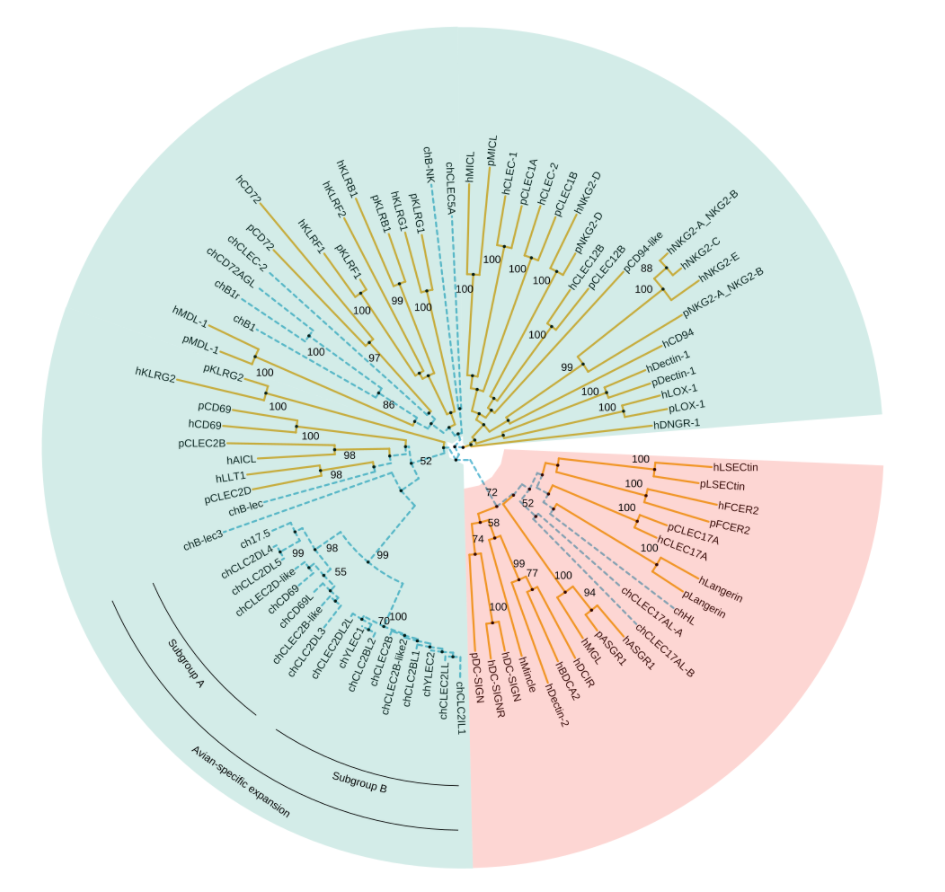
其他软件输出的结果
1 | |
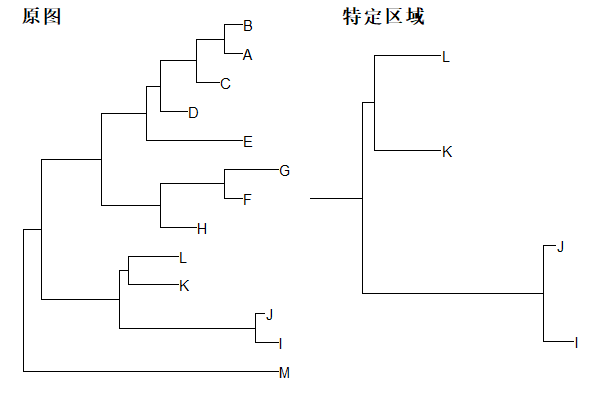
放大特定区域
1 | |
标准化选中的clade
1 | |
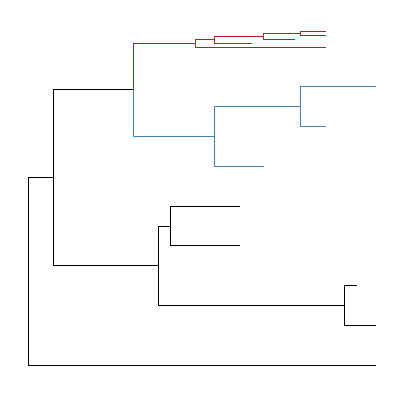
隐藏/展示某个clade
1 | |
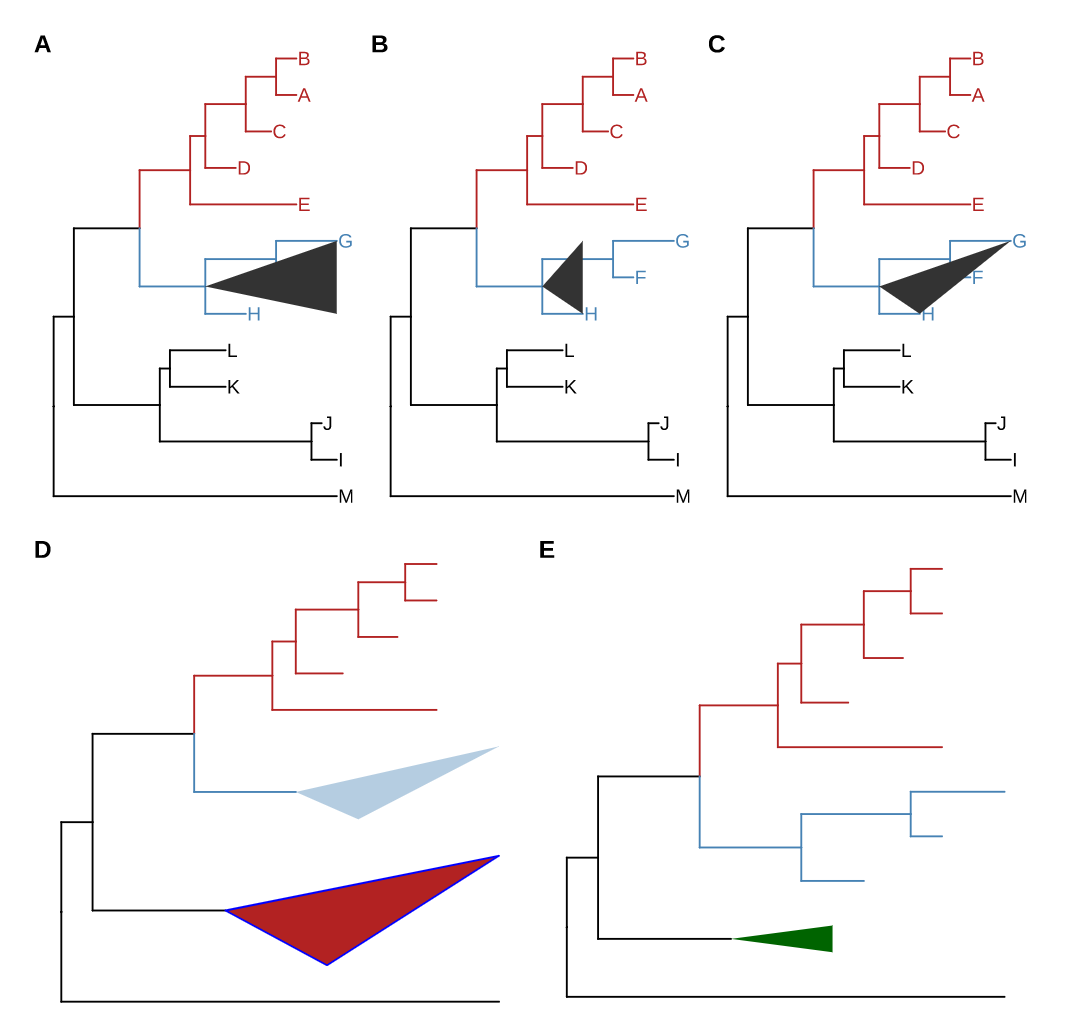
利用三角形隐藏/展示某个clade
1 | |
分组Taxa
1 | |
另外一种方法:
1 | |
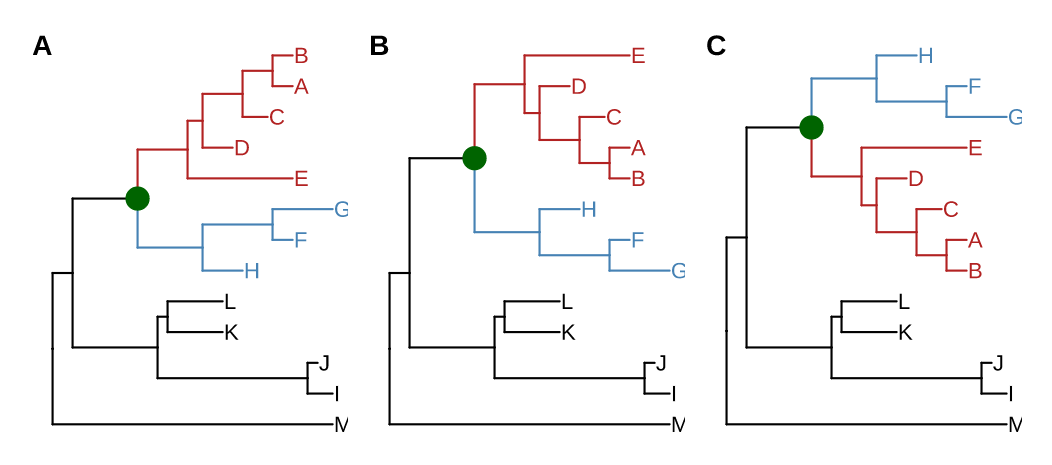
旋转clade
旋转clade有两种方法:
1 | |
外部数据mapping到树上
1 | |
将SNP数据添加到树上
1 | |
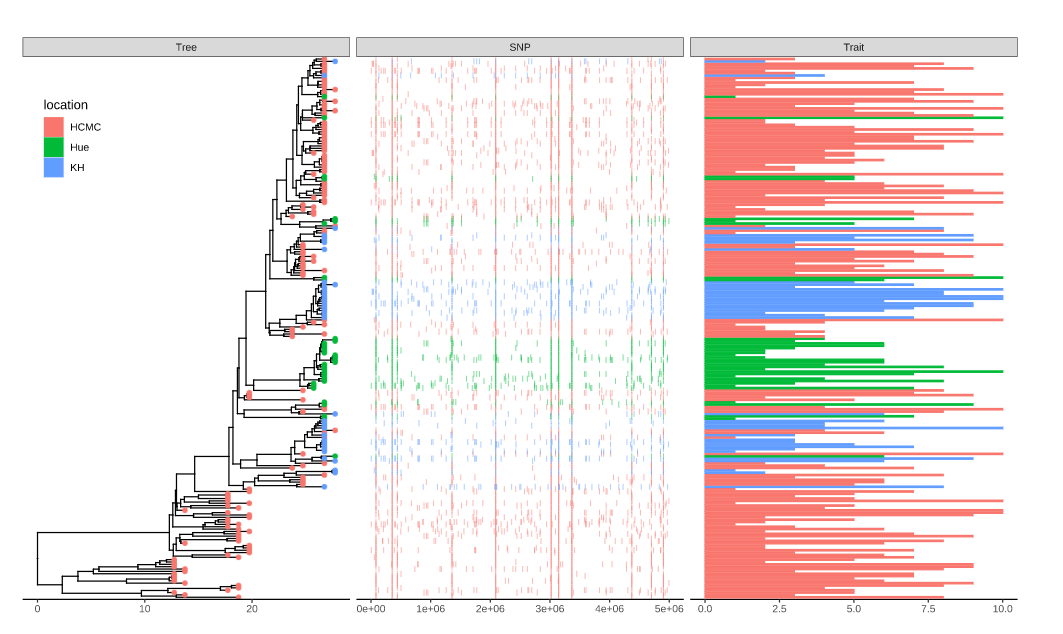
关联矩阵
1 | |
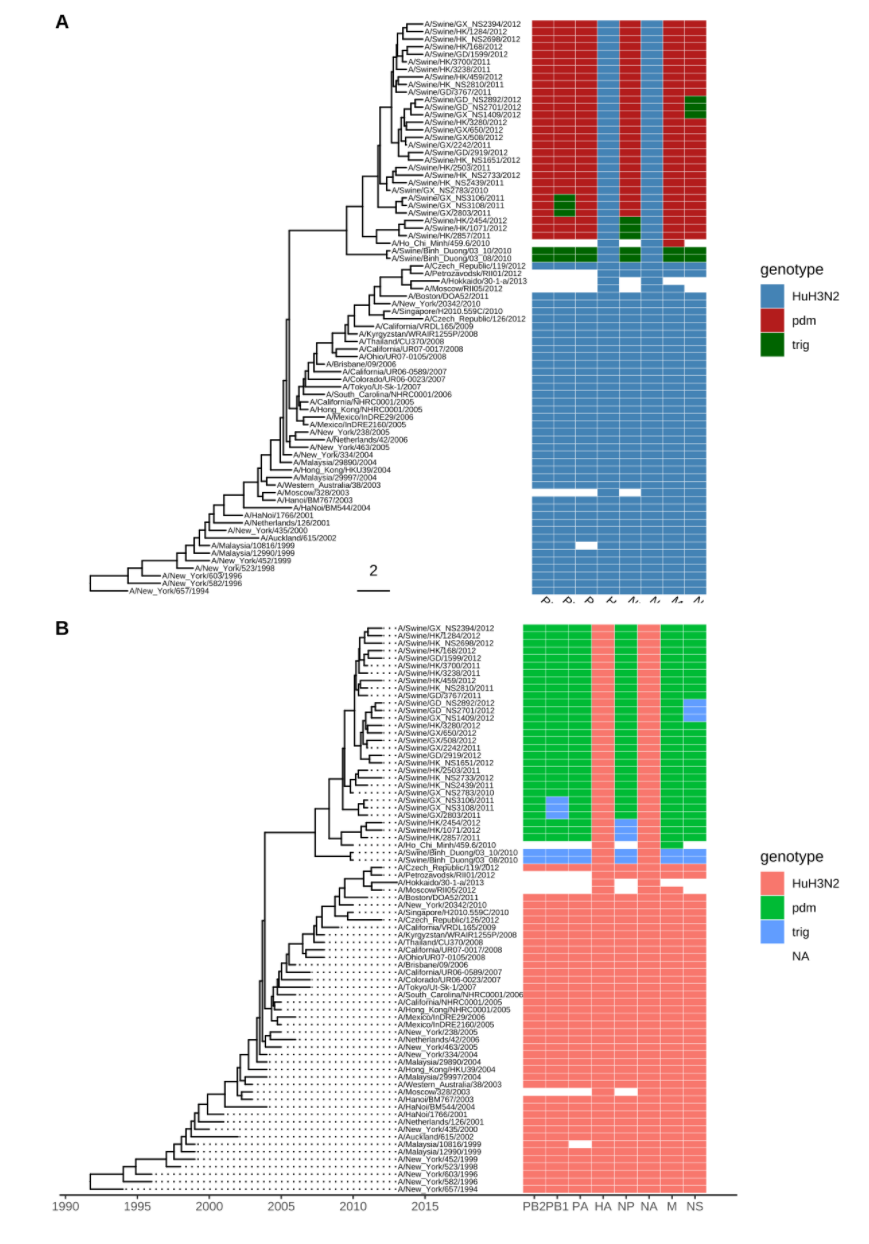
关联多个矩阵
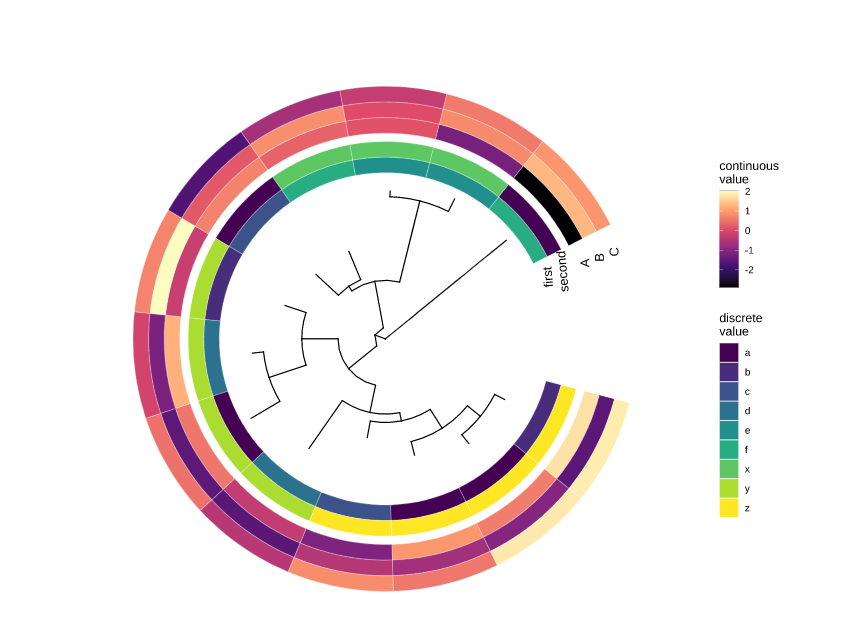
1 | |
多序列比对可视化
1 | |
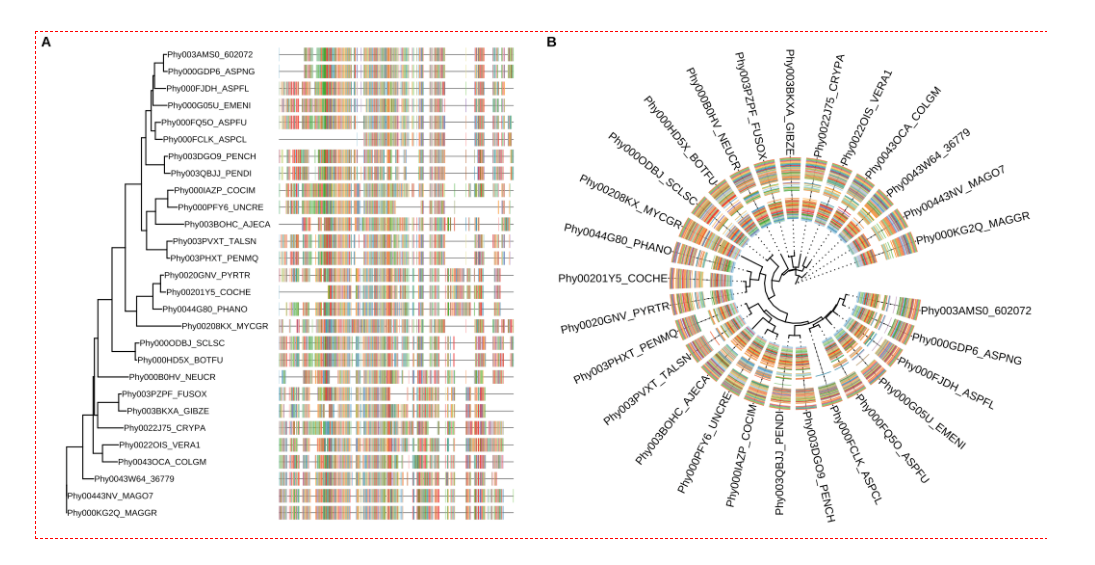
拼图
1 | |
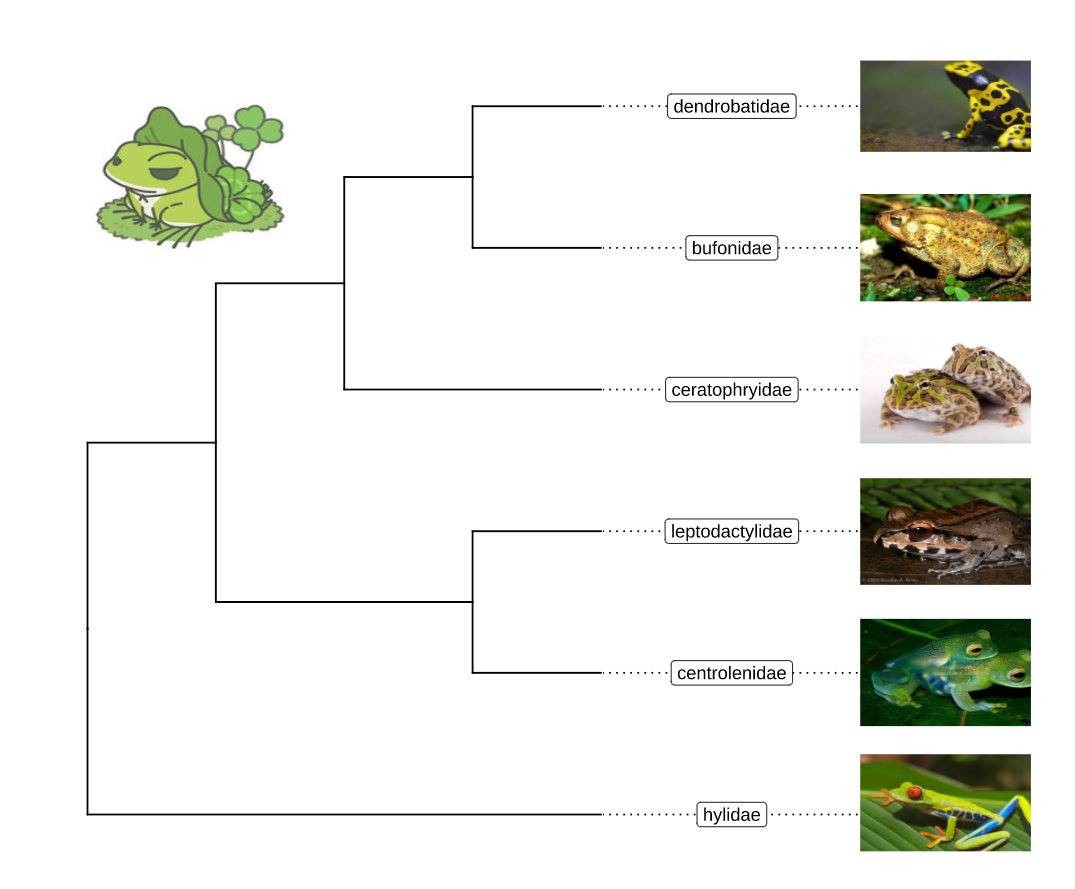
图片注释
没能找到图片下载地址,看代码很容易理解:
1 | |
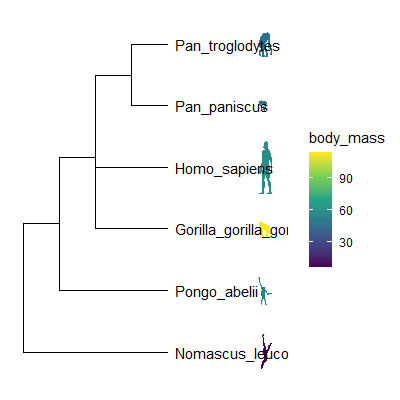
图标(剪影)注释
PhyloPic提供了1300余种生物剪影,ggtree能够调用这个数据库中的生物图标进行注释,相当于上面的图片。这种情况下绘图稍微有点慢,毕竟这个数据库是国外的。
1 | |
使用子图进行注释
用子图进行注释是通过函数geom_inset()来完成的。
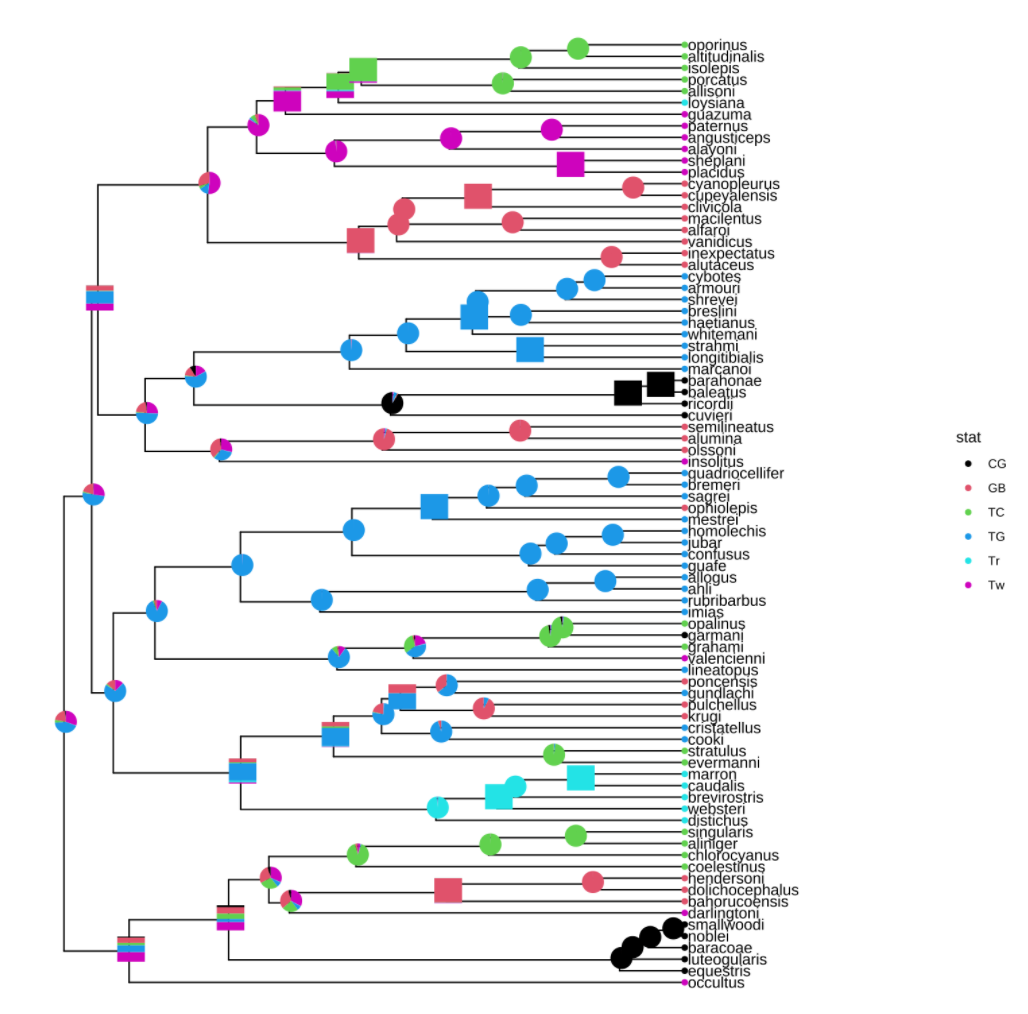
条形图注释
1 | |
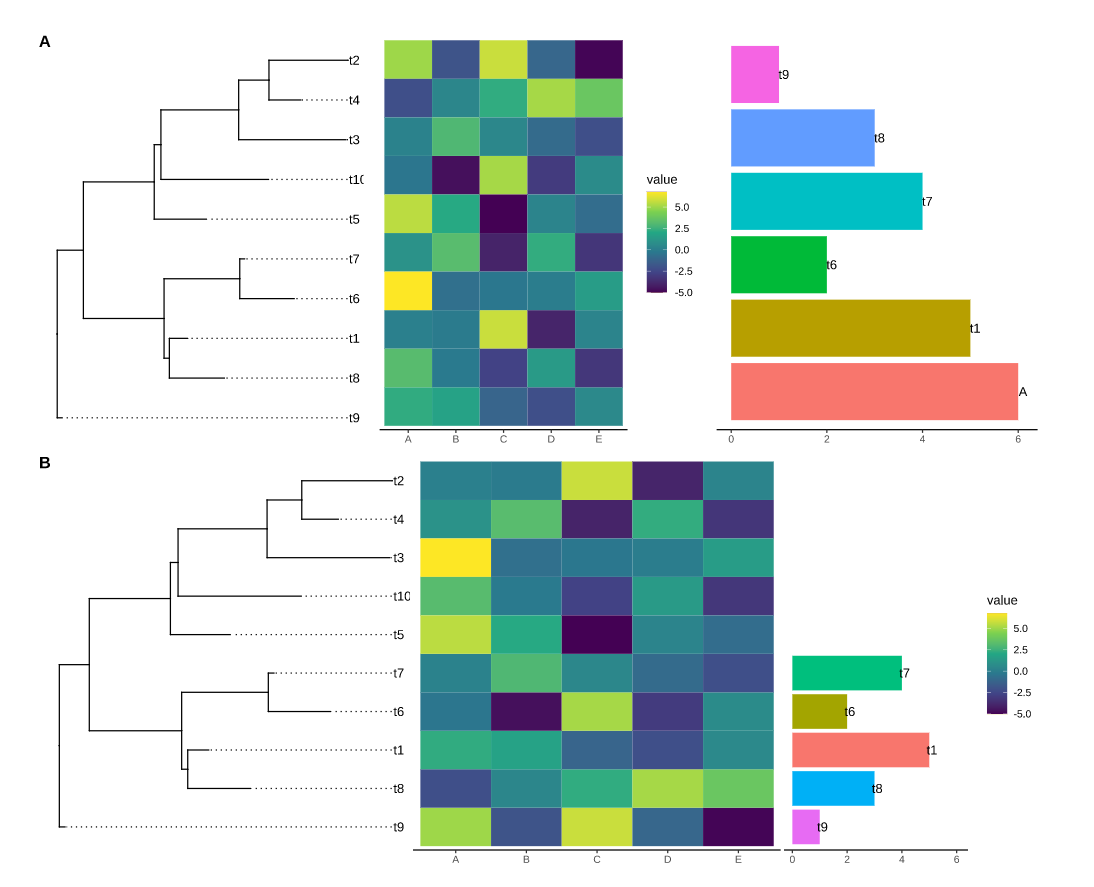
饼图注释
1 | |
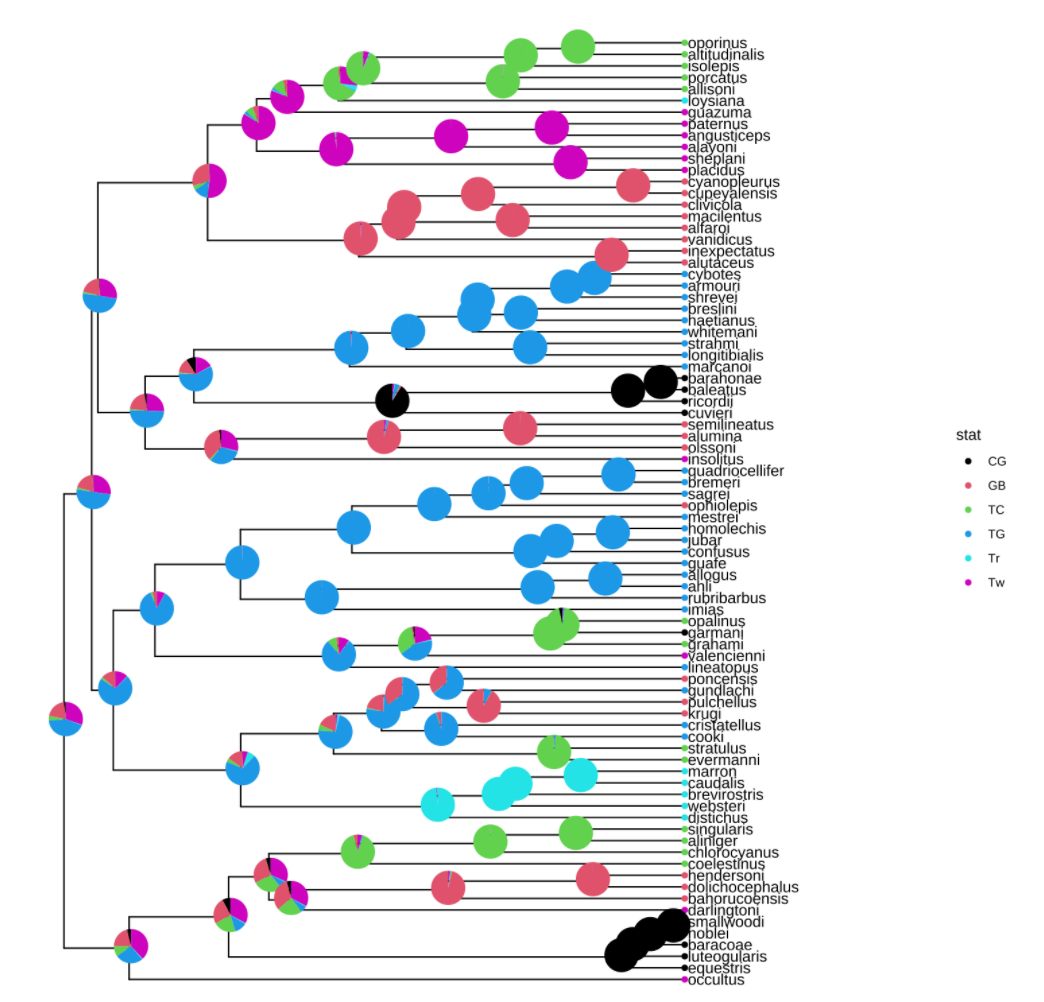
多种图像组合注释
1 | |
Phylomoji注释
1 | |
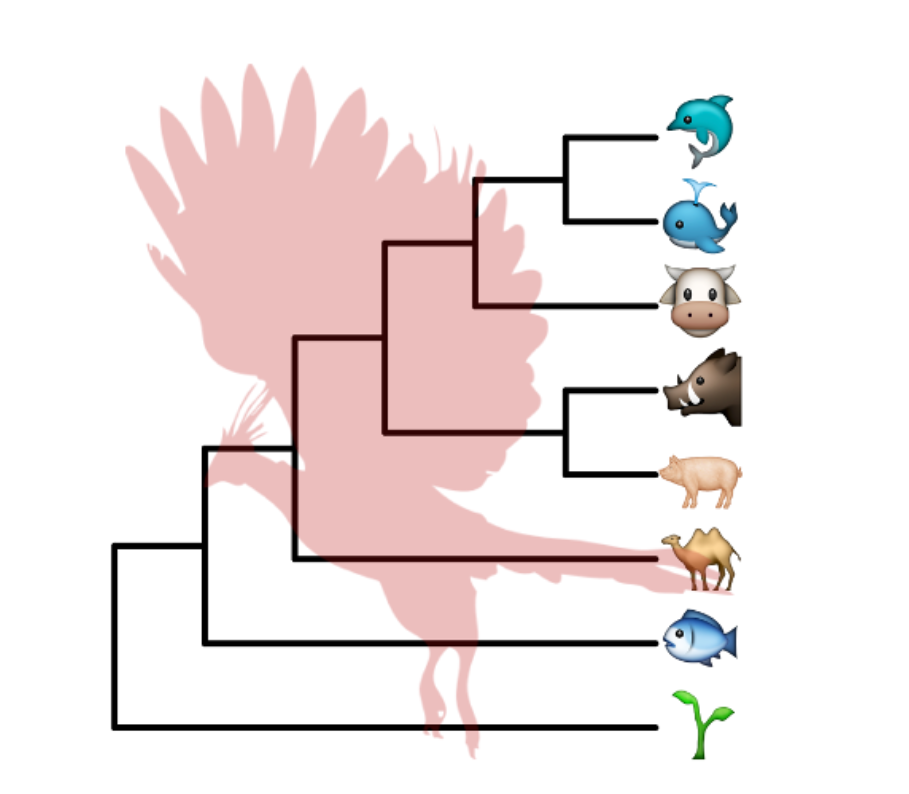
Emoji注释circular/fan树
1 | |

Emoji注释clades
1 | |
AppleColorEmoji
1 | |
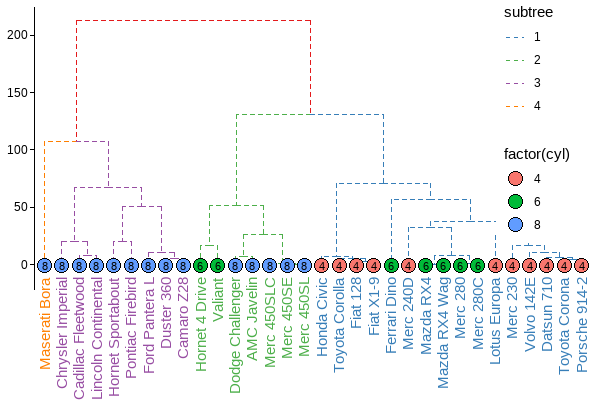
ggtree可视化其他数据
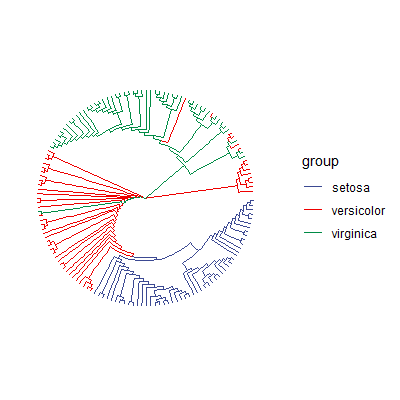
ggtree支持其他的数据类型,比如树状图(更多格式请移步Y叔博客):
1 | |
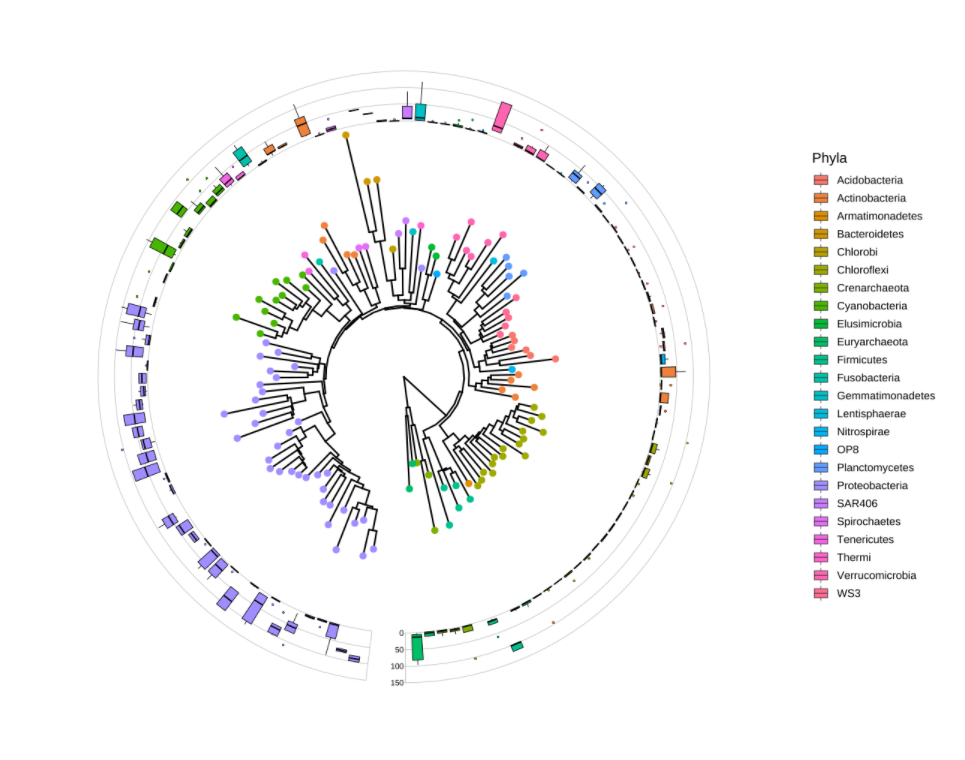
ggtree拓展ggtreeExtra
ggtreeExtra真的是惊艳到我了,只能献出我的膝盖啊!
ggtree的函数geom_facet()只支持rectangular、 roundrect、 ellipse 及 slanted 这4种输出样式,并不支持在circular、fan及radial这几种输出样式的外环上添加图层,为了解决这个问题,Y叔团队开发了新的R包:ggtreeExtra!
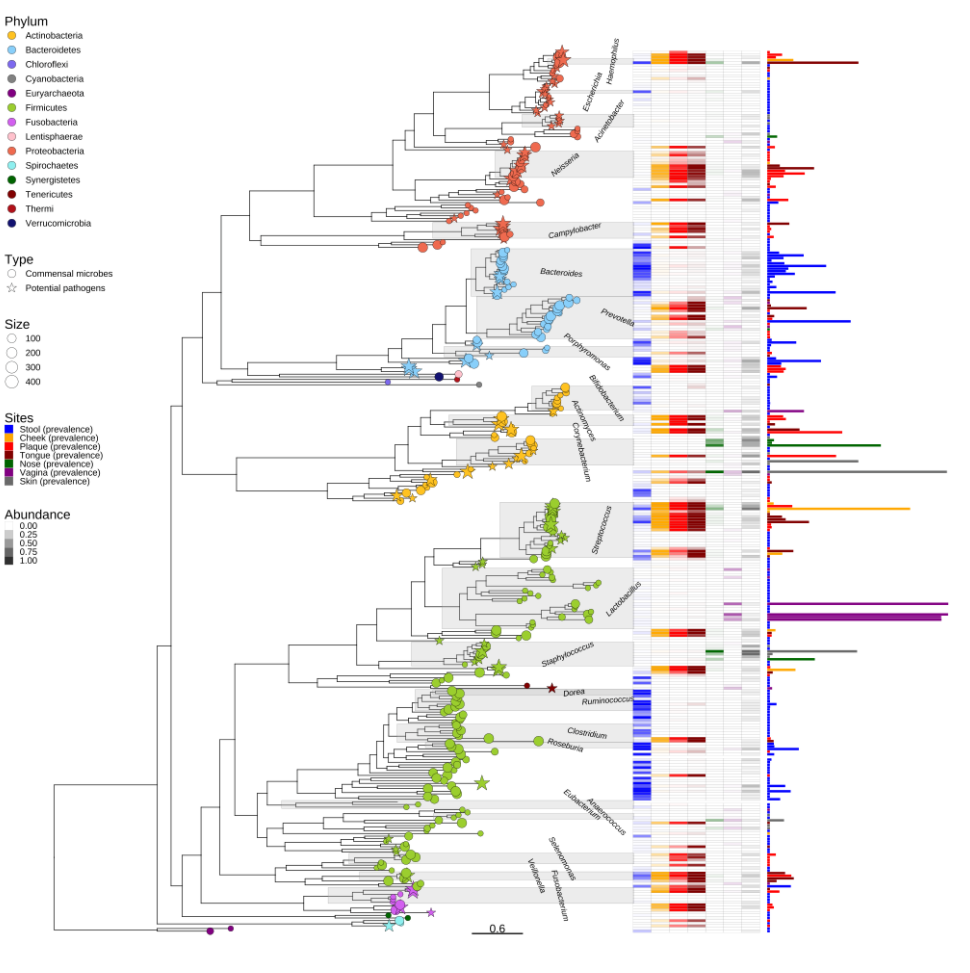
添加微生物组丰度
ggtree直接支持phyloseq对象,这个对做微生物的来说,简直就是福音啊!
1 | |
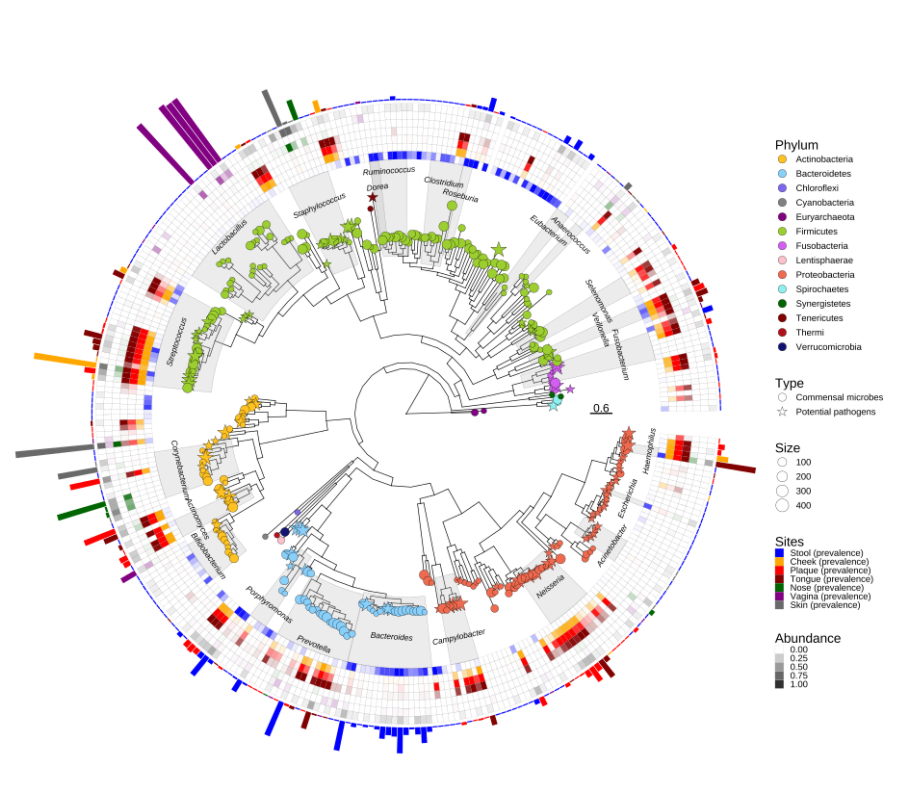
多维数据添加多个图层
1 | |
另外一种输出方式:
1 | |
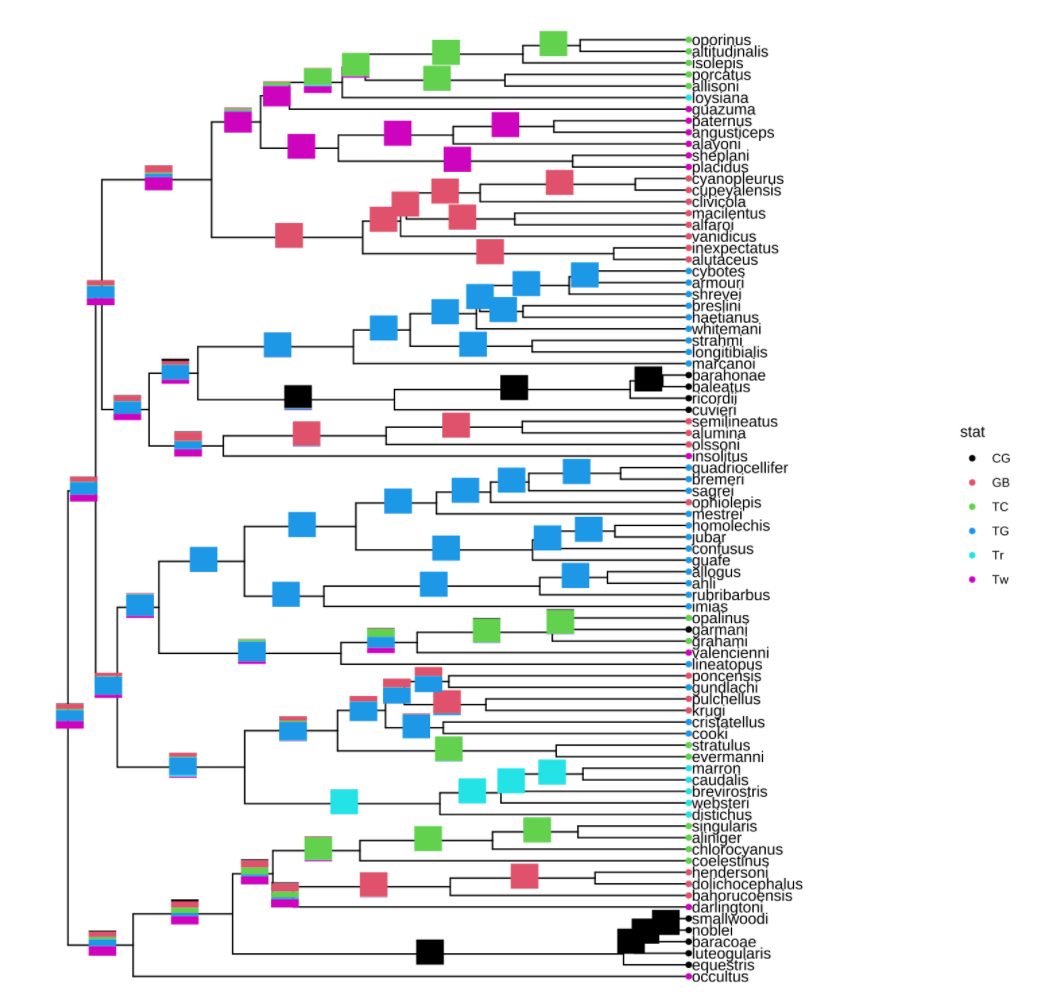
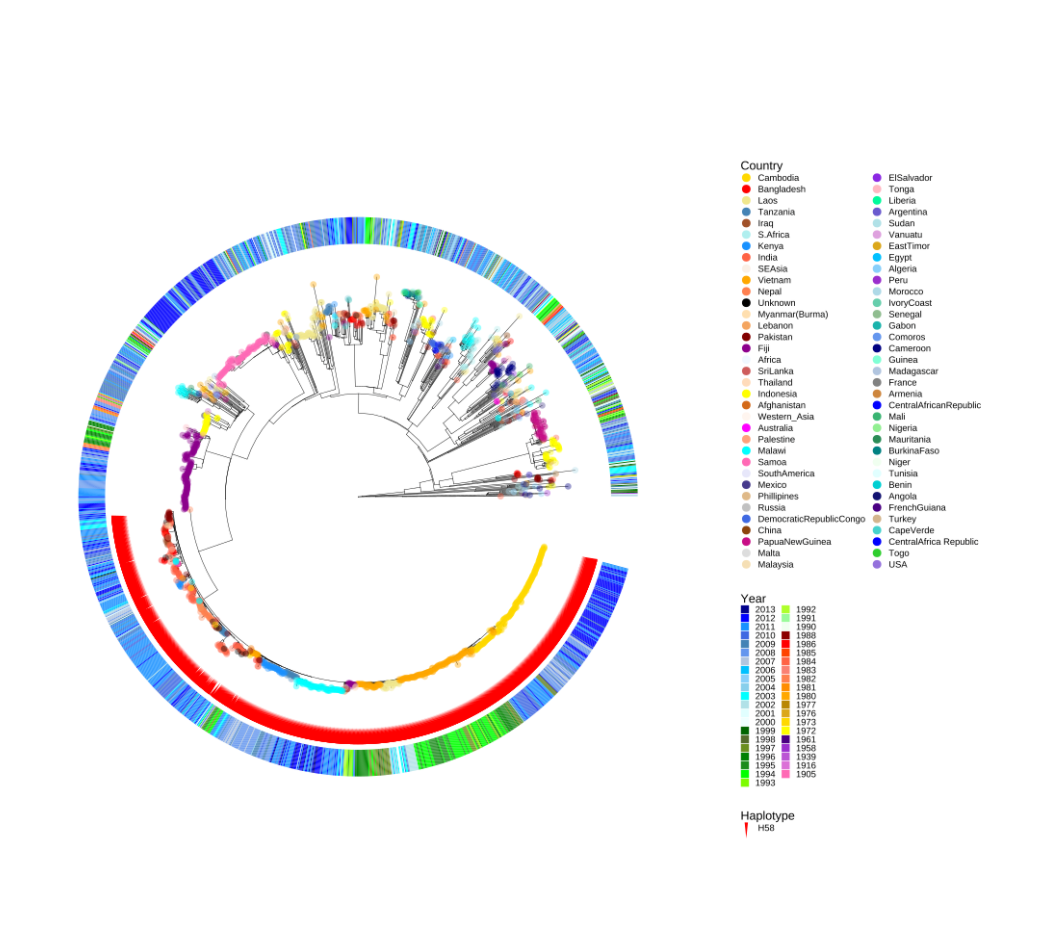
群体遗学传例子
1 | |

1 | |
参考文献
[1]. Yu G, Smith D K, Zhu H, et al. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data[J]. Methods in Ecology and Evolution, 2017, 8(1): 28-36.
[2]. https://yulab-smu.top/treedata-book/index.html
[4]. https://yulab-smu.top/treedata-book/chapter1.html
[5]. Wang L G, Lam T T Y, Xu S, et al. Treeio: an R package for phylogenetic tree input and output with richly annotated and associated data[J]. Molecular biology and evolution, 2020, 37(2): 599-603.
交流请联系:
💌lixiang117423@gmail.com
💌lixiang117423@foxmail.com