BRAKER学习笔记
1 BRAKER3的优势
- 可以使用转录组和蛋白数据
2 基因预测成功的关键
- 高质量的基因组:
short scaffolds太多的话不会得到很准确的结果。 - 简单的序列名称:如
Chr1这种是最好的。 - 要标记重复序列:
the genome should be masked for repeats,避免对重复序列和低复杂度区域预测到基因结构;转录组数据比对是重复序列也会影响;在GeneMark-ES/ET/EP/ETP和AUGUSTUS流程中,softmasking比hardmasking表现更好。softmasking:重复区域的核苷酸用小写字母,其他区域的用大写字母。hardmasking:重复区域的核苷酸序列用N表示。
- 注意分支特异性:例如部分真菌会有特殊的分支点等,需要根据物种信息选择合适的参数。
- 进一步使用预测结果之前需要确定预测的准确性。
3 BRAKER运行模式
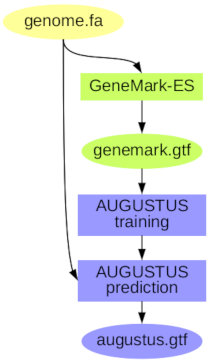
3.1 只有基因组
在这种模式下,GeneMark-ES使用基因组进行训练,GeneMark-ES预测的长基因用于训练后续的AUGUSTUS;最终使用AUGUSTUS进行预测。这种模式得到的结果的准确性是最低的。
3.2 基因组+转录组
短读长的RNA-seq数据适合这种预测方法。这种方法要求每个intro被很多的alignments覆盖,但是组装过的转录组比对不适用于这种方法。长读长的RNA-seq数据也是可以的,但是需要注意的是每个进行训练的转录本都需要多次测序并与基因组进行比对。现在BRAKER还不支持短读长和长读长的RNA-seq数据的混合使用。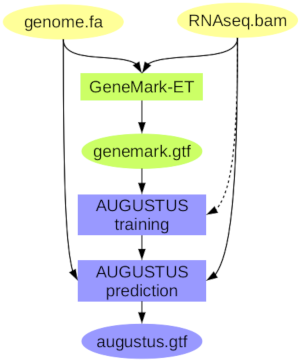
3.3 基因组+蛋白序列
这种方法尤其适用于没有RNA-seq数据的情况,而且使用的蛋白序列可以是来自和目标物种遗传距离未知的物种,当然,使用近缘物种肯定预测得更准确些。这个方法需要蛋白家族,也就是每个蛋白家族具有代表性的序列应该在数据库中。OrthoDB这个数据库是测试过的。官方提供了OrthoDB v.11和OrthoDB v.12.
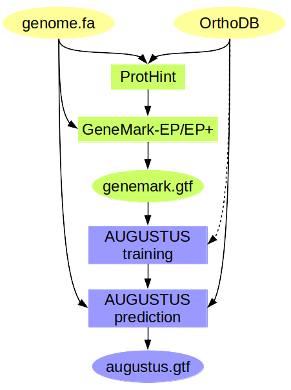
3.4 基因组+转录组+蛋白序列
从头预测+转录本+同源蛋白预测。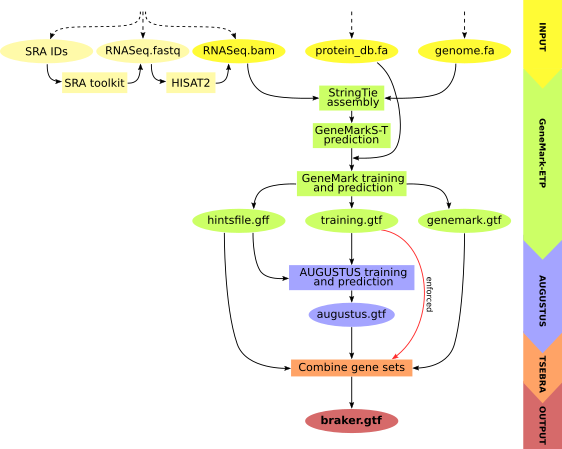
4 软件安装
4.1 Docker安装
推荐使用docekr进行安装:
1 | |
进入容器:
1 | |
4.2 Conda安装
如果没有docker权限,可以尝试使用conda进行安装:
1 | |
BRAKER和AUGUSTUS在conda上也是有的。个人推荐速度更快的Miniforge替代原生conda。
4.3 软件依赖
- AUGUSTUS 3.5.0 F2
- GeneMark-ETP (source see Dockerfile)
- BAMTOOLS 2.5.1R5
- SAMTOOLS 1.7-4-g93586edR6
- Spaln 2.3.3d R8, R9, R10
- NCBI BLAST+ 2.2.31+ R12, R13
- DIAMOND 0.9.24
- cdbfasta 0.99
- cdbyank 0.981
- GUSHR 1.0.0
- SRA Toolkit 3.00 R14
- HISAT2 2.2.1 R15
- BEDTOOLS 2.30 R16
- StringTie2 2.2.1 R17
- GFFRead 0.12.7 R18
- compleasm 0.2.5 R27
4.4 脚本依赖
主要的脚本是braker.pl,在运行braker.pl时会调用以下这些脚本: - `filterGenemark.pl
filterIntronsFindStrand.plhelpMod_braker.pmfindGenesInIntrons.pldownsample_traingenes.plensure_n_training_genes.pyget_gc_content.pyget_etp_hints.py
需要确保这些脚本都是可执行状态,也就是有x这个属性;如果不是可执行状态,需要使用chmod a+x *.pl *.py添加可执行属性。
5 运行BRAKER
建议使用softmasked Repeats后的基因组。5.1 模式选择
5.1.1 转录组
这个流程适用于有转录组但是没有蛋白质数据的情况。BRAKER有多种方式接收传入的转录组数据:- 使用
SRA数据:有多个SRA数据时使用,进行分隔,BRAKER会自动下载对应的SRA数据:1
braker.pl --species=yourSpecies --genome=genome.fasta --rnaseq_sets_ids=SRA_ID1,SRA_ID2 - 使用本地的
FASTQ文件:可以是为mapping过的原始数据,同样需要提供FASTQ文件的文件名称和对应的文件的路径,会根据文件的ID自动识别文件,如果是双末端的会识别为SRA_ID1_1.fastq和SRA_ID1_2.fastq这样,如果是单末端的会识别为ID.fastq.1
2
3braker.pl --species=yourSpecies --genome=genome.fasta \
--rnaseq_sets_ids=SRA_ID1,SRA_ID2 \
--rnaseq_sets_dirs=/path/to/local/fastq/files/ - 使用比对后的
bam文件1
2
3
4# 类似FASTQ输入方式
braker.pl --species=yourSpecies --genome=genome.fasta \
--rnaseq_sets_ids=BAM_ID1,BAM_ID2 \
--rnaseq_sets_dirs=/path/to/local/bam/files/
1 | |
使用bam文件时,BRAKER默认bam文件是HISAT2比对得到的结果;如果想使用SATR的话,需要加上参数--outSAMstrandField intronMotif.
- 从比对信息中提取的
gff格式信息:格式类似于这样:1
braker.pl --species=yourSpecies --genome=genome.fasta --hints=hints1.gff,hints2.gff第二列的1
2chrName b2h intron 6591 8003 1 + . pri=4;src=E
chrName b2h intron 6136 9084 11 + . mult=11;pri=4;src=Eb2h和最后一列的src=E是必须要的。
可以将上面这些方法进行组合,但是需要注意的是,如果使用转录组+蛋白组这种方式的话,不建议使用--hints这种方法。
5.1.2 蛋白组
这种方法尤其适用于没有转录组的情况。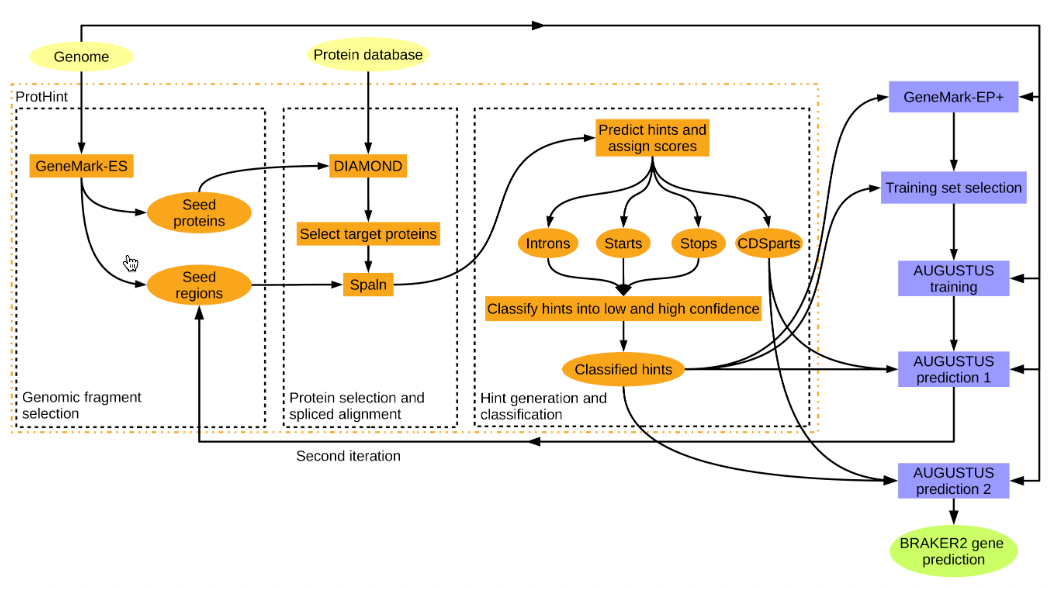
执行下面的代码即可运行:
1 | |
推荐使用
OrthoDB作为proteins.fa的基础。也可以使用
ProtHint的数据:1 | |
对应的文件是这样的:
1 | |
5.1.2.1 训练和预测UTRs
如果转录组只有bam格式文件,而且基因组是softmasked格式的,那么BRAKER就可以自动训练UTR的参数用于AUGUSTUS预测;预测完成之后就会使用覆盖度等信息进行基因预测,对应的命令是这样的:
1 | |
Warnings:
- 这个流程属于测试性的;
--UTR=on不兼容bamToWig.py这个脚本;--UTR=on会增加内存需求;- UTR预测有时能优化编码序列的预测准确度,但不是总是能够优化。
5.1.2.2 链特异性的转录组比对结果
对于UTR的训练和预测,链特异性的转录组数据能够提供更多的信息,将bam文件分割成+和-的两个文件,然后运行BRAKER:非链特异性的1
2
3braker.pl --species=yourSpecies --genome=genome.fasta \
--bam=plus.bam,minus.bam --stranded=+,- \
--UTR=onbam文件也可以用,不用于UTR预测,用于基因预测:1
2
3braker.pl --species=yourSpecies --genome=genome.fasta \
--bam=plus.bam,minus.bam,unstranded.bam \
--stranded=+,-,. --UTR=on5.1.3 转录组+蛋白组
首先使用转录组和蛋白组数据训练GeneMark-ETP,输出结果中属于high-confindent的基因用于训练AUGUSTUS,随后使用TSEBRA将这两个软件的结果进行整合。5.1.3.1 转录组比对
GeneMark-ETP使用Stringtie2对转录本进行组装,也就需要bam文件有XStag,如果比对的时候用的软件是HISAT2,那么就需要加上-dta这个参数;使用STAR的话就要加上--outSAMstrandField intronMotif这个参数选项。有几种代码运行方式:1
2
3braker.pl --genome=genome.fa --prot_seq=orthodb.fa \
--rnaseq_sets_ids=SRA_ID1,SRA_ID2 \
--rnaseq_sets_dirs=/path/to/local/RNA-Seq/files/
1 | |
1 | |
5.2 长/短读长转录组
5.3 长读长转录组+蛋白组
如果使用BRAKER的fast-hack模式,那么就需要使用长读长的转录组构造bam文件:
1 | |
然后拉取适合长读长转录组的
BRKAER版本:1 | |
运行:
1 | |
Warnings:
- 不能混合使用短读长和长读长的转录组数据。
isoseq数据的深度严重影响着基因预测的准确度。5.4 参数选择
5.4.1 —ab_initio
Compute AUGUSTUS ab initio predictions in addition to AUGUSTUS predictions with hints (additional output files:augustus.ab_initio.*. This may be useful for estimating the quality of training gene parameters when inspecting predictions in a Browser.5.4.2 —augustus_args=”—some_arg=bla”
知道物种适用的AUGUSTUS的话可以把这些参数传入,使用空格进行分隔,如--first_arg=sth --second_arg=sth.5.4.3 —threads=INT
指定线程数,某些步骤使用单线程,部分步骤使用多线程。设置超过8线程的话,对于多线程步骤会加速计算。5.4.4 —fungus
是GeneMark-ETP的参数选项,如果是真菌的话使用这个参数,其他不用。5.4.5 —useexisting
是否使用现成的配置文件,如果使用的话就会覆盖掉软件默认的参数文件。5.4.6 —crf
是否为AUGUSTUS执行CRF training,生成的参数只是用于最后的预测,使用的话会增加运行时间。5.4.7 —lambda=int
默认的是参数值是2,如果有大量单个外显子的基因的话,参数可以设置为0.5.4.8 —UTR=on
见上述步骤。5.4.9 —addUTR=on
见上述步骤。5.4.10 —stranded=+,-,.,…
见上述步骤。6 输出结果
braker.gtf:最终的基因集,不同的模式有不同的结果ETPmode:AUGUSTUS和GeneMark-ETP的整合结果otherwise:官方不建议使用的模式和结果。
braker.codingseq:最终基因集的编码序列。braker.aa:最终基因集的蛋白序列。braker.gff3:最终基因集的注释文件,需要加上--gff3这个参数选项。Augustus/*:Augustus的输出结果。GeneMark-E*/genemark.gtf:GeneMark-ES/ET/EP/EP+/ETP的输出结果。hintsfile.gff:从转录组或者是蛋白质组中输出的结果。braker_original/*:BRAKER输出的结果,这个结果是运行compleasm之前的结果。bbc/*:best_by_compleasm.py的输出结果。
如果设置--skipGetAnnoFromFasta参数的话不会生成编码序列和蛋白序列结果。
7 测试练习
7.1 数据下载
基因组数据来自文章A pangenome reference of wild and cultivated rice,选择的是品种是GP119,这个基因组是soft masking的,下载地址RicePandb.

蛋白质组数据来自Rice Resource Center的R498这个水稻品种。
转录组数据来自文章的提供的数据:
- 使用biohelpers中的
get_fq_meta下载测序数据的meta信息:这四个文件是GP119的转录组数据:1
2
3get_fq_meta -id PRJEB73710 -o PRJEB73710.meta.txt
grep GP119 PRJEB73710.meta.txt | grep TRANSCRIPTOMIC | awk '{print $1}'1
2
3
4ERR13336195
ERR13336317
ERR13336603
ERR13336454 - 使用biohelpers中的
get_fq_file下载测序数据的meta信息(全部下载后再筛选)1
get_fq_file -id PRJEB73710 -m save -t ftp -o ./
1 | |